All published articles of this journal are available on ScienceDirect.

Solitary Radiolucent Erdheim-chester Disease: A Case Report and Literature Review
Abstract
Background:
Erdheim-chester disease (ECD) is a rare non-Langerhans histiocytosis of unknown etiology, which typically presents with bilateral symmetric osteosclerosis and multi-organ involvement. Lesions may be intraosseous or extraosseous and involve the heart, pulmonary system, CNS, and skin in order of decreasing likelihood.
Objective:
The objective of this study is to discuss a case of erdheim-chester disease and conduct a review of the literature.
Case:
We describe a rare case of erdheim-chester in an asymptomatic 37-year-old male who was diagnosed after suffering a right ulnar injury. Subsequent evaluation revealed a solitary radiolucent ulnar lesion without multi-system involvement.
Results & Conclusion:
The case is unique in its solitary distribution, lytic radiographic appearance, and asymptomatic presentation preceding pathologic fracture. This presentation may simulate multiple other bone lesions.
1. INTRODUCTION
Erdheim-chester disease is a very rare non-Langerhans histiocytosis with less than 500 reported cases in the literature. The mean age of onset is 53 years with slight male predominance [1]. Clinical presentation varies depending on site of involvement, with many patients having osseous involvement and one or more extra-osseous sites [2]. Diagnosis is based on pathology and immunohistochemistry. The most common bone manifestation is mild, persistent juxta-articular pain, usually in the lower limbs, knees, and ankles. Patients almost universally have bilateral symmetric osteosclerosis of the long bone diaphyses and metaphyses with subchondral involvement reported more rarely. Typical lesions show symmetric cortical thickening and skeletal scintigraphy will demonstrate lesional increased uptake. ECD can be difficult to diagnose since it is a very rare disease that can affect multiple organ systems simultaneously and requires a multidisciplinary approach. The current case presented is unique even among ECD cases due to its solitary distribution, lytic radiographic appearance, and completely asymptomatic course prior to pathologic fracture.
2. CASE PRESENTATION
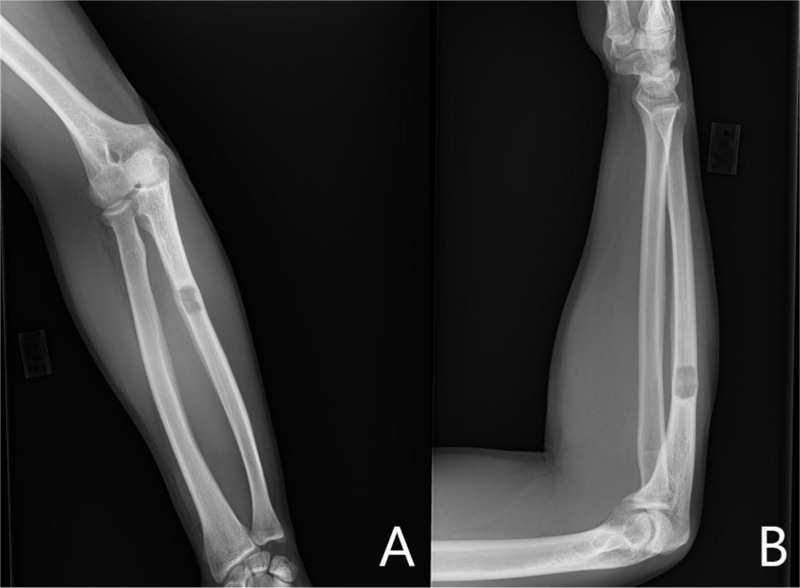
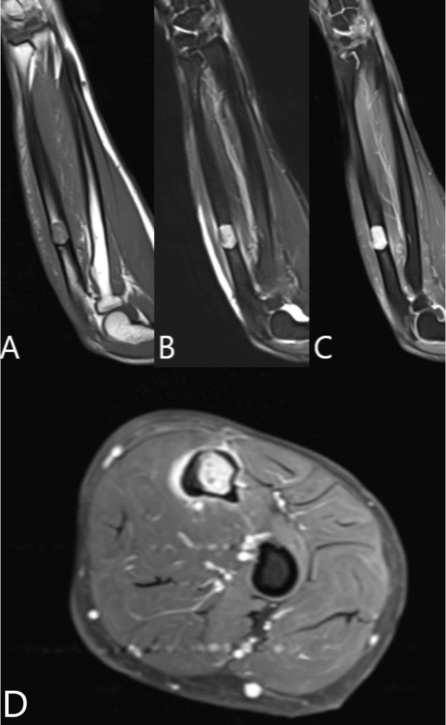
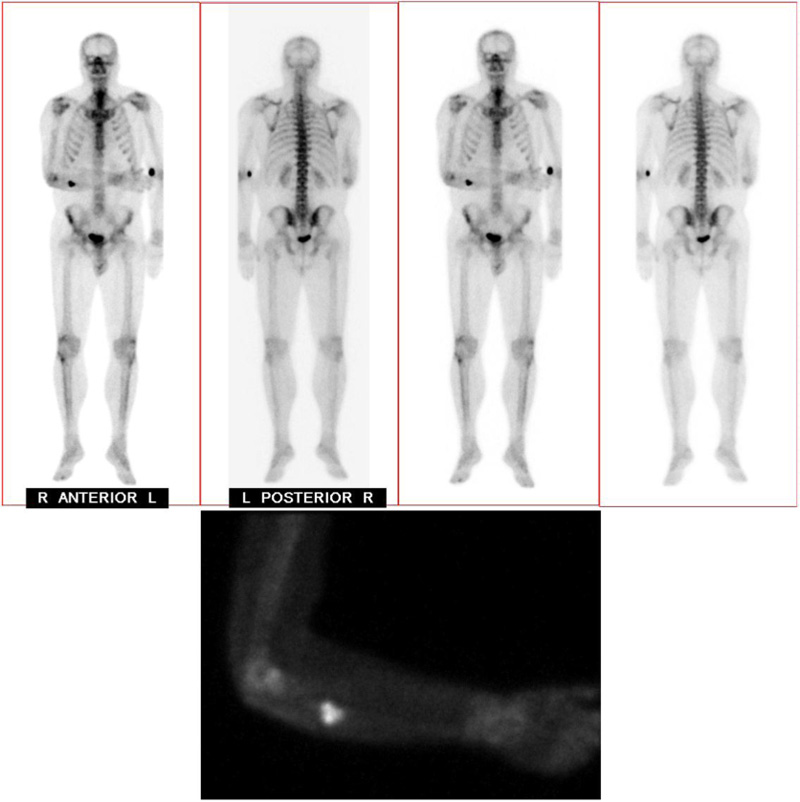
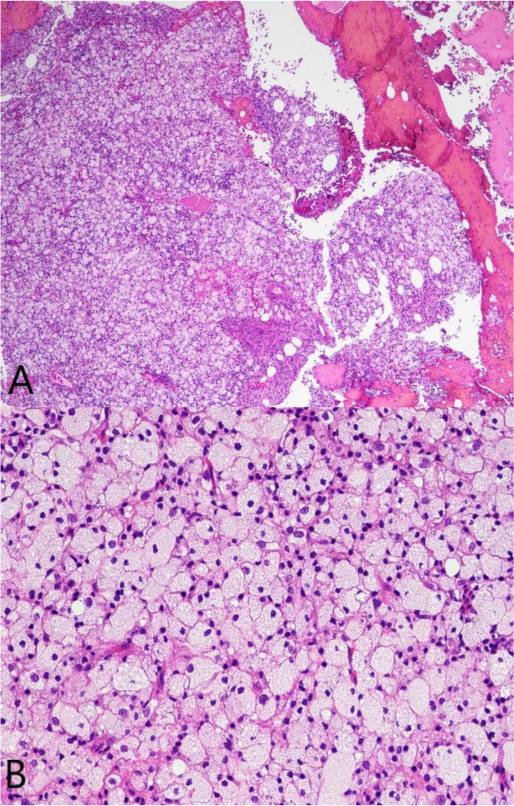
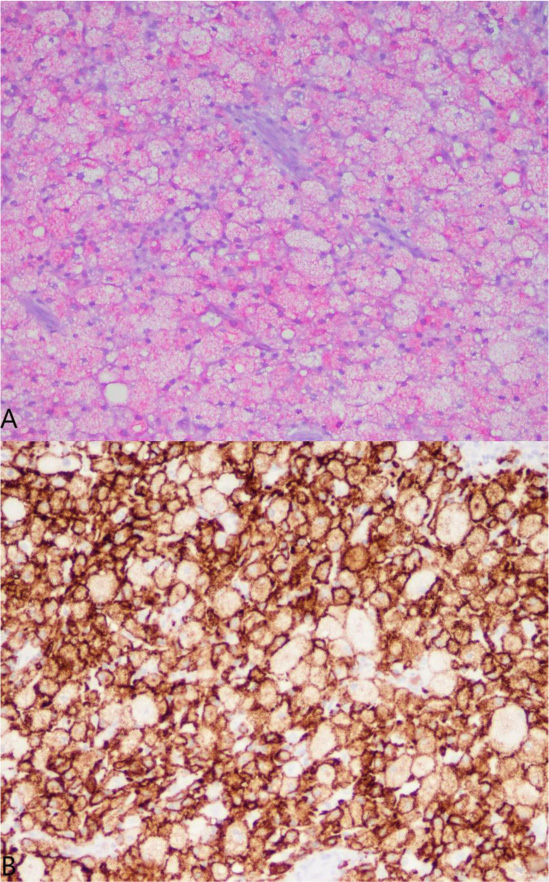
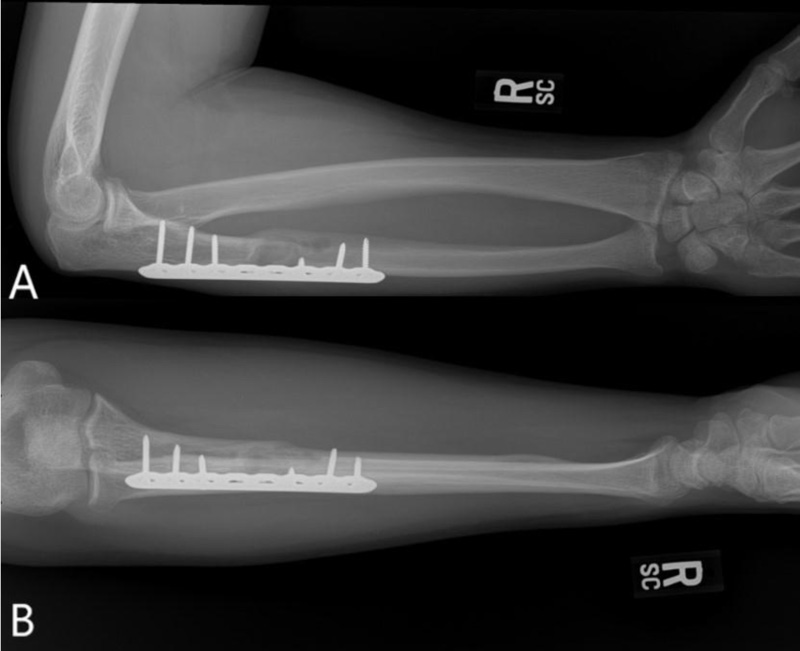
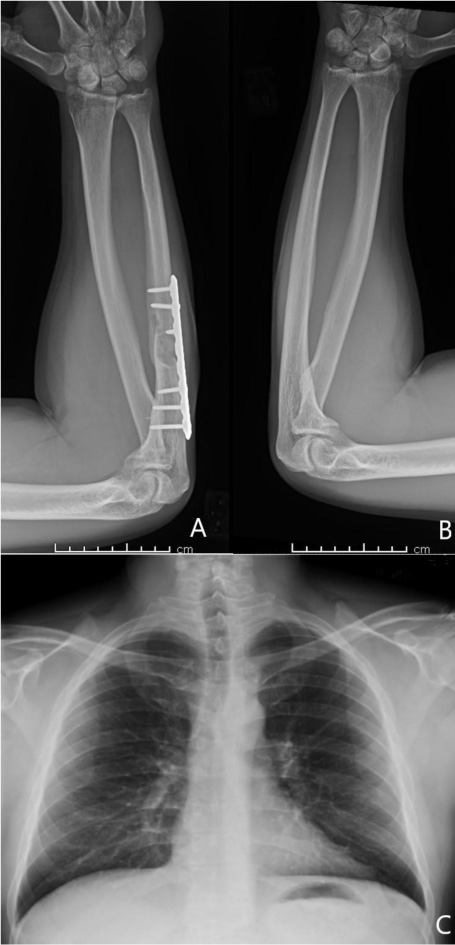
A 37-year-old male with no significant medical history suffered a fall onto his outstretched arm, leading to immediate right forearm pain, causing him to seek medical attention. He denied any prior history of right upper extremity pain, prodromal pain, or other musculoskeletal symptoms. His family history and medical history were unremarkable. Radiographs of the right upper extremity showed a minimally-displaced fracture of the ulnar diaphysis through a geographic, central purely lytic lesion (Fig. 1). The patient was treated initially with splint immobilization and referred to an orthopedic oncologist. Upon arrival to our hospital, the patient was informed that data concerning his case might be submitted for publication and the patient consented. MRI of the right forearm was obtained before and after administration of intravenous contrast (Fig. 2). A well-circumscribed, slightly heterogeneous solid lesion was found within the medullary space of the proximal right ulnar diaphysis. No notable intraosseous perilesional edema was noted, but there was extraosseous edema consistent with a recent fracture. Nuclear medicine Tc99 whole body skeletal survey demonstrated solitary increased uptake in the right ulnar lesion (Fig. 3). Increased periarticular activity within the right elbow joint was suspected to represent increased articular blood flow. Surgery was performed consisting of open biopsy, curettage, and open reduction internal fixation of the right ulna (Fig. 4). An intraoperative frozen section indicated a benign fibrous lesion with foamy histiocytes. Subsequently, the true histiocytic nature of the infiltrate and diagnosis was confirmed with positive CD68 and CD163 immunostains (Fig. 5). However, the histiocytes were negative for S100 and CD1a, excluding Rosai-Dorfman disease and Langerhans cell histiocytosis, respectively (Table 1). A PAS-diastase test was partially positive in the histiocytes, but no rod-shaped intracellular bacteria were identified. A gram-stain was negative as well, excluding Whipple’s disease. Although foamy histiocytes may be seen with fracture, the one-month gap between fracture and surgery and dense sheet-like replacement of bone marrow by histiocytes suggested a true neoplastic pathology rather than a reactive process. All of these findings indicated ECD. Although half of ECD cases have V600E BRAF mutations, this specimen was negative [3]. Due to common visceral involvement by ECD, the patient was referred to medical oncology for further evaluation. Follow-up plain radiography bone survey did not indicate additional sites of bone involvement. MR brain images, CT imaging of the chest, abdomen, and pelvis, and an echocardiogram were normal. It was concluded that the patient had a case of solitary bone-only ECD. Multidisciplinary consultation between medical oncology, radiation oncology, and orthopedic oncology agreed upon serial surveillance with bone surveys and clinical examinations every 6 months. At the latest orthopedic oncology follow-up 6 months postoperatively, the patient was doing well and returned to all activities without restriction. Skeletal survey and forearm radiograph remained negative for lesions, and the ulna showed progressive fracture healing with graft incorporation (Figs. 6 and 7).
| Name |
Most Common Site of Involvement |
Lesion Appearance | S100 | CD1a | CD68 | Factor XIII | BRAF V600E | Incidence |
|---|---|---|---|---|---|---|---|---|
| ECD | Metaphysis and diaphysis of long bones | Osteosclerotic | Negative | Negative | Positive | Positive | >50% of cases positive | Unknown; 500 cases reported |
| Rosai-Dorfman | Lymph nodes | Indurated papules | Positive | Negative | Positive | Negative | Negative | Unknown; 650 cases reported |
| Juvenile Xanthogranuloma | Skin | Red/yellow papules, plaques or nodules | Negative | Negative | Positive | Positive | Rarely positive | Less than 0.5% of pediatric tumors |
| Kikuchi Disease | Lymph nodes | Yellowish, nectrotic foci | Positive | Positive | Positive | Negative | Negative | Unknown |
3. DISCUSSION
Histiocytic disorders are derived from mononuclear phagocytic cells and can be subdivided into Langerhans cell histiocytosis, non-Langerhans histiocytosis and malignant histiocytic disorders. Non-Langerhans histiocytoses are derived from the monocyte-macrophage lineage (Table 1). All have an unknown incidence, but even the most prevalent one, Juvenile Xanthogranuloma, represents less than 0.5% of pediatric tumors and histiocytoses [4]. erdheim-chester disease (ECD) is a non-Langerhans histiocytic disorder typified by multifocal osteosclerotic lesions of the long bones in addition to organ infiltration. ECD is quite rare, with fewer than 500 cases reported, but exact prevalence is unknown [5]. It has been diagnosed in all age groups, most commonly in adults, with slight male predominance [1]. While symptoms vary, most clinical presentations include localized bone pain, diabetes insipidus, neurologic, and constitutional symptoms. The most frequent site of involvement is the long bones, of which there is typically ubiquitous bilateral symmetric osteosclerosis. Most patients present with persistent juxta-articular pain, usually in the lower extremities. Less commonly, patients are asymptomatic and incidentally diagnosed with radiography for unrelated reasons [6]. Cardiac involvement, varying from valve defects to conduction defects to periaortic fibrosis, is also present in the majority of ECD patients [7, 8]. There may also be pulmonary, retroperitoneal, CNS, and skin involvement [9-12]. The current case is unique in its solitary bone only distribution and osteolytic radiographic appearance rather than the typical bilateral, symmetric, and osteosclerotic presentation with organ involvement.
Various imaging modalities are used to both locally and systemically stage ECD. Plain radiography and Tc99 scintigraphy are vital for determining the extent of bone involvement, as the skeleton is affected in 96% of cases [13, 14]. Bone scans characteristically reveal intense bilateral areas of osteoblastic activity, particularly affecting the diaphyses and metaphyses [15]. MRI is useful to determine the extent of bone involvement and to rule out osteonecrosis, which may result from the disease itself or as a consequence of treatment [13]. Although less commonly used, CT of extremities often exhibits bilateral bone lesions [16]. Other conditions such as Paget’s disease or metastases must be excluded. However, symmetrical scintigraphy is unique to ECD, unlike both Paget’s and metastases. This further illustrates the uniqueness of our patient. F-labeled fluorodeoxyglucose PET/CT scans can also be used for staging, especially as a biomarker for BRAF mutations [17].
Since cardiovascular involvement is seen in 75% of cases with death resulting in 60% of cases, echocardiography is crucial for assessment at diagnosis [13]. ECG will be non-specific, however, showing only generalized conduction defects [8]. Pericardium thickening is the most common cardiovascular finding, followed by plaque-like tissue encircling the aorta [18].
Brain MRI is also important as CNS involvement is present in 51% of cases. MRI shows diffuse hypointense regions on T1W in either a meningeal or infiltrative pattern, variable in T2W images [19, 20]. Orbital involvement has also been reported and demonstrated by either MRI or CT(10). Optic nerve compression was noted with dysfunction of extra-ocular muscles in some cases and others progressing to blindness [21, 22].
Within the visceral organs of the abdomen and pelvis, ECD manifests as a mass-like infiltrative process [23]. In patients with localized abdominal involvement, MRI and CT will show perinephric infiltration (67%), renal sinus expansion (56%), renal artery stenosis (49%), and periaortic infiltration (43%). The perinephric infiltration results in a spiculated appearance and has been termed the “hairy kidney sign”, pathognomonic of ECD [24]. Presence of these abdominopelvic findings is significantly associated with a positive BRAF mutation [25]. BRAF mutation is associated with F-FDG-avid CNS disease, greater standardized uptake values (SUVs) within lesions, and greater mortality rates [3, 26].
Histology exhibits sheets of xanthomatous histiocytes derived from the myeloid lineage interspersed with inflammatory and multinucleate giant cells within surrounding fibrosis [27]. Due to its rarity and varying manifestations, ECD must be distinguished from other histiocytic and dendritic cell disorders. Langerhans cell histiocytosis (LCH) is another osteolytic histiocytic disease that can involve multiple sites but most commonly affects the bones. Skin manifestation is more common in LCH, and the two diseases can be distinguished based on morphology and immunohistochemistry. Cell markers necessary for diagnosis of ECD include CD68, CD163 and Factor XIIIa, whereas markers of LCH include CD1a and S100 [28-30]. In contrast to LCH, CD1a and S100 are negative in ECD [31]. The BRAF V600E mutation is another important distinguishing factor from other non-Langerhans cell histiocytoses, as half of the patients with ECD have the mutation [32]. The BRAF gene is a proto-oncogene of the Raf kinase family of growth signal transduction protein kinases and it regulates the MAP kinase (MAPK) and ERK signaling pathways. In its non-mutated state, the BRAF gene regulates cell division and differentiation through the incorporation of extracellular signaling. However, when mutated, growth signals proceed in the absence of any extracellular signaling, thereby leading to unregulated cell growth and survival. The V600E mutation is the most common BRAF mutation that leads to constitutive activation and unregulated signaling through the MAPK/ERK pathways. In addition, more than 30 other mutations of the BRAF gene exist, most of which also affect BRAF activation [33-35]. Testing for the V600E mutation can be done by RT-PCR, immunohistochemistry via VE1 monoclonal antibody, pyrosequencing and/or Sanger sequencing [36]. These methods are highly sensitive and specific. However, the BRAF V600E mutation is not specific to ECD, as it may also be present in LCH, papillary thyroid carcinoma, hairy cell leukemia, and melanoma [37-41].
Treatment of ECD is reserved for symptomatic disease, CNS involvement, and evidence of organ dysfunction [42]. Symptomatic patients with a BRAF mutation should receive treatment with a BRAF inhibitor. Patients with the mutation have a better prognosis as they can be administered the B-Raf inhibiter Vemurafenib, resulting in an 86% 2-year progression-free survival rate [32, 43]. Symptomatic patients without BRAF mutation are recommended pegylated interferon alfa or MEK inhibitors [44]. Interferon-alfa non-responders are recommended cladribine and cyclophosphamide [45, 46]. In our case, with isolated bone involvement, only orthopedic treatment via open reduction and internal fixation of the fracture with curettage and grafting have been necessary. Serial surveillance will continue.
CONCLUSION
Erdheim-chester disease is a very rare non-Langerhans histiocytosis with less than 500 reported cases in the literature. Patients almost universally have bilateral symmetric osteosclerosis of the long bone diaphyses and metaphyses with lesions demonstrating cortical thickening. However, ECD is very difficult to diagnose since it is an extremely rare disease that may simultaneously affect multiple organ systems. Our patient’s case is unique due to its solitary distribution, lytic radiographic appearance, and asymptomatic presentation preceding pathologic fracture. Medical oncology, orthopedic oncology, and radiation oncology all agreed upon serial surveillance with bone surveys and clinical examinations every 6-months, illustrating the need for a multidisciplinary approach towards staging and treatment of ECD.
LIST OF ABBREVIATIONS
| CT | = Computed tomography |
| ECD | = Erdheim Chester Disease |
| ECG | = Electrocardiogram |
| LCH | = Langerhans Cell Histiocytosis |
| MRI | = Magnetic Resonance Imaging |
| RT-PCR | = Reverse Transcription Polymerase Chain Reaction |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
Not applicable.
CONSENT FOR PUBLICATION
The patient provided consent for participation and publication.
STANDARDS OF REPORTING
All work in this manuscript was written according to the CARE Guidelines for case reports.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Aleksandar Popovic – Literature review, writing, editing, paper organization Dr. Christopher Curtiss – Writing, editing, histopathological specimen analysis Dr. Timothy A. Damron – Writing, editing, paper organization.

