All published articles of this journal are available on ScienceDirect.

Primary Hyperparathyroidism in the Common Orthopaedic Practice
Abstract
An extensive review of the publications on primary hyperparathyroidism (pHPT) is presented in this report. It has strongly been emphasized in the literature that patients with pHPT may present either with the classical symptomatology or with asymptomatic disease, emerged due to biochemical screening. The clinical and epidemiological presentation of pHPT in western countries has changed profoundly during the past few decades, and bone disease is nowadays a distinct rarity. The introduction of serum calcium screening for osteoporosis and the technological advances in the laboratory assessment of parathyroid hormone have played important roles in early diagnosis. Subsequently, the disease is increasingly being detected as asymptomatic hypercalcaemia without guiding signs or symptoms. A third type of disease, the normocalcaemic variant, has been recently described in the literature. However, the potential diagnosis of pHPT should always be on the orthopaedics’ list of differential diagnoses in female or elderly patients with vertebral fractures and nephrolithiasis, either symptomatic or asymptomatic, as well as when solitary or multiple osteolytic lesions are encountered on the radiographs.
Additionally, a middle aged woman with parathyroid adenoma and subsequent brown tumors detected on the pelvic radiographs is reported. Her initial laboratory findings indicated a minimal increase of the serum calcium, a mild increase of the erythrocyte sedimentation rate, and a significant increase in total serum alkaline phosphatase. Finally, the detection of elevated parathyroid hormone levels indicated the diagnosis of pHPT and necessitated imaging studies of the parathyroid glands, which indicated a parathyroid adenoma. Following successful excision of the parathyroid adenoma, the patient suffered from the hungry bone syndrome. After a follow-up of 20 years, the patient had normal calcium, vitamin D, and parathyroid hormone serum levels, while a pelvic radiograph indicated no significant changes in the appearance of the brown tumors.
1. INTRODUCTION
The purpose of this report is to perform an extensive review of the relevant international literature about primary hyperparathyroidism (pHPT), to indicate the value of clinical suspicion for the early diagnosis of the disease in a patient presenting in the common orthopaedic practice, and to present an illustrative case with a 20-year follow-up.
2. REVIEW
2.1. Parathyroid Hormone
The parathyroid hormone, also called parathormone (PTH), is secreted by the chief cells of the four parathyroid glands. It is a cornerstone of calcium homeostasis through its increased calcium absorption effect in the gastrointestinal tract, kidney, and the skeleton by osteoclasts. It also plays a role in activating vitamin D. It is a polypeptide containing 84 amino acids, whose action is opposed by the hormone calcitonin. The PTH has both anabolic and catabolic skeletal effects that vary according to the kinetics of serum levels and the type of bone. The anabolic effects are manifested in patients with a periodic, rapid, and transient rise in serum PTH, as seen with the daily subcutaneous injection of recombinant human PTH (1-34) and PTH (1-84) in the treatment of osteoporosis. These patients have increased Bone Mineral Density (BMD) at skeletal sites with a high trabecular component, such as the lumbar spine, and a reduction in fracture risk. The catabolic effects are typified in patients with chronic, persistently elevated PTH levels.
2.2. Hyperparathyroidism
Hyperparathyroidism (HPT) is due to increased activity of the parathyroid glands either from an intrinsic abnormal change altering excretion of PTH (primary and tertiary HPT) or from an extrinsic abnormal change affecting calcium homeostasis stimulating the production of PTH (secondary HPT). Secondary HPT (sHPT) may result from a primary response to a physiological stimulus, such as in calcium malabsorption syndromes, chronic renal disease, or chronic hypocalcaemia following vitamin D deficiency, particularly in elderly people. Hyperparathyroidism may persist in a significant proportion of patients with sHPT and chronic kidney disease after kidney transplantation, a state known as tertiary HPT.
All types of HPT have many symptomatic features and skeletal manifestations in common. From a biochemical perspective, their main difference is that pHPT causes an increase in serum calcium and reduces phosphate, while sHPT produces the opposite [1-4].
2.2.1. Primary Hyperparathyroidism
2.2.1.1. Introduction
Primary hyperparathyroidism is one of the most common endocrine diseases. It is characterized by the autonomous, incompletely regulated, excessive secretion of PTH. It is most commonly sporadic. It is caused by a solitary parathyroid gland adenoma, usually affecting a single parathyroid gland, in 80-85% of the patients, and in 15-20%, it is associated with all four-gland parathyroid hyperplasia or multiple adenomas in hereditary or familial syndromes, while less than 1% is due to carcinoma. The female to male ratio is approximately 3:1. It is usually diagnosed in postmenopausal women, although younger patients and women entering menopause may also be involved. Three forms of the disease have been described so far. It may appear with the typical symptomatology or with atypical clinical findings, while a third type, the normocalcaemic variant, has recently been discovered. It is the commonest cause of hypercalcaemia (excluding humoral hypercalcaemia of malignancy). The disease is traditionally diagnosed by hypercalcaemia associated with elevated PTH serum levels, a critical finding for the differentiation of pHPT from diseases or tumors associated with hypercalcaemia. Hypercalcaemia is defined as raised total and ionized serum calcium concentrations; severe hypercalcaemia is associated with acute signs and symptoms, while mild hypercalcaemia is often asymptomatic. Normocalcaemic pHPT is an uncommon condition, a forme fruste of the disease. It is defined by normal total and ionized serum calcium concentrations but with PTH levels that are consistently elevated in the absence of secondary causes of HPT. The elevated PTH should be detected by the more recent assays that detect the 1-84 full-length molecule [5-24].
2.2.1.2. Clinical Presentation
Several studies have suggested that pHPT may be associated not only with the classical renal stones and bone manifestations but also with non-classical ones, such as cardiovascular, gastrointestinal, myotendinous, articular and neuropsychological symptoms, impaired quality of life, and increased cancer risk. Patients usually present with systemic symptoms of HPT or local clinical findings of related skeletal lesions. General early symptoms usually include fatigue, weight loss, difficulty in concentrating, somnolence, muscle weakness, hyperreflexia, and polyuria-polydipsia. Patients affected by pHPT are often misdiagnosed because their dominant picture may include nonspecific symptoms due to severe hypercalcaemia, such as gastritis, constipation, and depression, leading to diagnostic confusion. In severe symptomatic pHPT, renal involvement is manifested by hypercalciuria, nephrolithiasis, and nephrocalcinosis. The disease is also cited as one of the causal diseases that induce secondary osteoporosis. Osteoporosis and hypercalciuria associated with vertebral fractures and nephrolithiasis, respectively, may occasionally be asymptomatic. Nonetheless, the routine use of imaging and of biochemical determinations have revealed the frequent occurrence of asymptomatic reduced kidney function, in terms of estimated glomerular filtration rate, as well as increased morbidity and mortality, which has been defined as chronic kidney disease. In elderly patients, pHPT is more frequently asymptomatic and mostly characterized by bone involvement, while younger patients are more often symptomatic and mainly affected by renal impairment [25-36]. Data from the international literature have revealed the increased correlation of urine calcium excretion with the risk of stone formation. Moreover, calcium excretion is significantly greater in younger patients with pHPT [37-42]. Patients with normocalcaemic pHPT have more substantial skeletal involvement than is typical, and they develop more findings and complications over time [43]. It has been emphasized in the world publications that pHPT should always be included in the differential diagnosis of patients with a history of nephrolithiasis, nephrocalcinosis, osseous pain, osteoporosis-osteopenia, and subperiosteal bone resorption, as well as in patients with a personal history of neck irradiation, lithium therapy or a family history of multiple endocrine neoplasia syndrome [4, 44]. Recent observations have suggested an association between vitamin D deficiency and severity of pHPT, indicating that the disease seems to be more severe in those with concomitant vitamin D deficiency, while vitamin D deficiency and insufficiency seem to be more prevalent in patients with pHPT than in geographically matched populations [45-50].
During the past few decades, the clinical presentation of pHPT has changed globally towards an oligo- or asymptomatic disease with uncertain and long-lasting progression, while most patients have minimal overt symptomatology with no renal or skeletal manifestations. Since the frequent, routinely performed measurement evaluation of serum calcium levels in developed countries using automated multichannel analyzers and the development of assays for intact 1-84PTH, the diagnosis is usually made during osteoporosis screening or fortuitously in asymptomatic patients. Many publications have shown that the incidence increases with age and is higher in women [51-54]. In contrast, paediatric pHPT is considered a very rare endocrinopathy. It is frequently associated with pathological fractures; therefore, it may be easily misdiagnosed as osteogenesis imperfecta [55, 56].
2.2.1.3. Bone Disease
The chronic excessive hypersecretion of PTH in pHPT has a significant impact on bone remodeling. Parathyroid hormone stimulates both bone resorption and bone formation. Markers of the increased bone turnover include bone formation markers (serum alkaline phosphatase, serum bone alkaline phosphatase, and serum osteocalcin), bone resorption markers, and longitudinal bone turnover marker studies. Alkaline phosphatase level, the most widely clinically available marker, can be mildly elevated but in many patients is within normal limits. In addition, bone densitometry, bone histomorphometry, and newer non-invasive imaging techniques offer insights into the skeletal manifestations of pHPT. Bone turnover increases by about 50%, leading to increased resorption at the endosteal envelope, increased cortical porosity, and thinning of cortical bone with a decrease in BMD. Bone density, as measured by photon absorptiometry and bone histomorphometry, shows a deficit of cortical bone and preservation or increase in cancellous bone elements in mild pHPT with no clinical evidence of skeletal disease. Bone loss may also be readily appreciated by densitometry, showing a typical pattern in which cancellous bone of the lumbar spine is reasonably well preserved, whilst the cortical bone of the distal third of the radius is preferentially reduced. The latter is a relatively uniform finding and is associated with an increase in forearm fracture risk. Bone densitometry is advised, particularly for monitoring bone mass at the midradius or femoral neck, in patients with pHPT. Most densitometry studies support the concept that the PTH appears to be catabolic at cortical sites and may have anabolic effects at cancellous bone sites [57-64]. However, some studies have indicated that the overall fracture risk is increased in untreated pHPT patients at both peripheral sites and in the spine. The reported increase in vertebral fracture risk does not fit with the observation that cancellous bone mass is preserved in pHPT patients [65-70]. Many of these patients may have a multifactorial risk profile for osteoporosis and bone fractures [71]. The preservation of bone mass demonstrated in long-term follow-up studies of patients with mild pHPT may be due to the fact that, despite the cortical bone loss, the cancellous bone structure remains unchanged or even improved, which may offset the cortical bone loss. In severe cases of pHPT, the negative effects on cortical bone may override the positive impact on cancellous bone, leading to bone loss and increased fracture risk [72]. An alternative explanation for the apparent preservation of trabecular bone is the fragmentation of the cortex by intracortical remodeling. The cortical fragments resemble trabeculae and so may be erroneously included in the quantification of trabecular bone density [73].
The risk of pathological fractures is of concern at many sites, such as the vertebrae, the distal forearm, the ribs and the pelvis, while there is no evidence of risk for hip fractures. However, fractures of the vertebrae and appendicular bones seem uncommon in modern pHPT, even in the long-term follow-up [74-89]. Finger fractures may be an early skeletal manifestation of pHPT, especially when they occur in young adults, in the absence of major trauma [90, 91]. In cases with delayed bone union of a mechanically stable fracture, once an infective cause has been excluded, other causes of the delayed union, such as pHPT, should be ruled out [92-94]. Rupture of a muscle or tendon [95-100] and intraarticular calcification [101] may also be evident. In addition, pHPT should be included in the wide variety of endocrine imbalances complicated by slippage of the upper femoral epiphysis. Worldwide, only nine cases of slipped capital femoral epiphysis have been reported associated with pHPT [102-106].
2.2.1.4. Osteitis Fibrosa Cystica
Bone disease in severe, long-lasting pHPT is described classically as Osteitis Fibrosa Cystica (OFC) or von Recklinghausen disease of the bones. The clinical signs and symptoms of OFC usually include generalized or focal bone and joint pain, acute radicular symptoms at the lumbar spine, and skeletal deformities secondary to pathological fractures. The radiographic features include subperiosteal bone resorption of the radial border of the phalanges, osteopenia, distal tapering of the clavicles, the typical salt-and-pepper skull demineralization, and brown tumors. Although historically considered a classical finding, OFC is nowadays a very rare manifestation of pHPT, identified in less than 2% of patients with pHPT, due to the early diagnosis of the disease. However, even in cases with no evidence of OFC, the skeleton is already affected [107-120].
Brown tumors may be due to a parathyroid gland adenoma or carcinoma. Rarely, they may be the first symptom of primary, secondary or tertiary HPT. They are reported in approximately 3% and 1.5% of patients with pHPT and sHPT, respectively. They are caused by excessive activity of osteoclasts. The increased rapid bone resorption leads to haemorrhage, proliferating fibrous cyst, and reparative granulation tissue formation with haemosiderin deposition. Their name originated from their gross histological appearance being a brownish mass. They present as a slow growing palpable bone swelling and may cause pain or pathological fractures when the brown tumor corrupts over two thirds of the cortex of a long bone, especially in the weight-bearing area. Brown tumors may also often be incidental findings on radiographs for another indication. They most likely occur in the pelvis, the ends of long bones, shoulders, mandible, ribs, clavicles, and rarely in the skull. They are not a true tumor, being an unusual reactive benign nonneoplastic process due to bone remodeling from either HPT or paraneoplastic syndrome. Their clinical, radiographic, and histological features share many similarities with other primary and secondary bone tumors. When they occur as a single lesion, the differential diagnosis may include reactive lesions (intraosseous haemorrhage, haemophiliac pseudotumor), benign lesions (giant cell tumor/osteoclastoma /tumor gigantocellularis, giant cell reparative granuloma, aneurysmal bone cyst, nonossifying fibroma, chondroblastoma), and malignant tumors. In cases presenting with multiple lesions, differential diagnosis may include metastatic tumors or multiple myeloma. Malignant tumors can also produce similar symptoms as part of the paraneoplastic syndrome that is due to the secretion of high levels of PTH-related Peptide (PTHrP), mimicking the effect of PTH on bones. Radiographic features and Computed Tomography (CT) scans may reveal a single or multiple osteolytic lesions. Their appearance may be complicated due to the involvement of both cortical and trabecular microstructure by abnormal osteoclastic activity. They may be relatively well-defined and sometimes surrounded by a sclerotic rim. They may be multilobular or associated with bone expansion, various bone erosion, cortical attenuation and potential pathological fractures. A malignant lesion should be considered in the differential diagnosis of ill-defined, mixed lytic/sclerotic lesions, in lesions with no margins, and in brown tumors with soft tissue involvement. In these cases, histological confirmation is recommended, although diagnosis may be challenging [121-142]. On Magnetic Resonance Imaging (MRI), the lesion is hypointense on T1-weighted images and hypointense or hyperintense on T2-weighted images. A cystic lesion is characterized by a watery signal. The enhanced scanning is obviously strengthened, and intratumoral haemorrhage leads to fluid-fluid level appearance, which has been referred to as a typical feature of an aneurysmal bone cyst but has also been reported in many other haemorrhagic osseous lesions. The final definitive diagnosis mainly depends on histological findings of the biopsied bone lesion, as neither cellular atypia nor mitotic figures are observed in brown tumors [143-146]. Management of the brown tumors consists mainly treating the HPT, often leading to spontaneous regression of the brown tumors. Surgery may be considered if large bone defects with spontaneous fracture risk or increasing pain are present. Tumor curettage, plombage with bone cement, and less invasive stabilization systems have proved to be acceptable surgical options [127, 147, 148].
2.2.1.5. Parathyroid Imaging
Parathyroid gland imaging including ultrasound, 99mTc-sestamibi (the drug consists of the radioisotope 99mtechnetium bound to sesta or 6, methoxyisobutylisonitrile ligands), scintigraphy, four-dimensional CT, and single photon emission CT fused with CT images (SPECT/CT) may have an important complementary role in the localization of the tumor as well as the detection of ectopic parathyroid tissue in a wide variety of unsuspected anatomic locations. Parathyroid gland imaging is useful for the localization of pHPT but not for the diagnosis of this entity [149-162].
2.2.1.6. Treatment
Parathyroidectomy is the only definitive treatment for pHPT. As more patients present with oligo- and asymptomatic pHPT or with a slow progression of the disease, ideal treatment has become more controversial, requiring the consideration of safe, nonoperative management in a selected group of patients. These patients may require intervention for the management of osteoporosis and/or hypercalcaemia. On the other hand, accumulated evidence confirms that the majority of patients with mild pHPT suffer from vague, nonspecific complaints that usually improve following parathyroidectomy [11]. Parathyroidectomy is definitely indicated for those with the symptomatic disease as well as for those with the asymptomatic disease who have evidence of end-organ sequelae. A key component in the decision to perform parathyroidectomy on patients with pHPT is their skeletal status [163-165]. Furthermore, parathyroidectomy has been demonstrated to improve BMD and reduce bone loss at both the lumbar spine and the femoral neck, reduce fracture risk and nephrolithiasis, improve health-related quality of life, and possibly increase overall survival [166]. On the other hand, a study performed 3 years following surgical removal of the parathyroid adenoma indicated that bone turnover decreased markedly, as judged from biochemical and histomorphometric data, but no changes were seen in trabecular bone structure. In addition, the relative cortical width increased and the cortical porosity decreased, while the bone mineral content of both cancellous and cortical bone areas increased. A decrease in the biochemical markers of bone formation and bone resorption has also been detected following treatment [98]. Therefore, all patients with pHPT should be referred for a potential surgical treatment by an experienced endocrine surgeon. When a patient also requires surgical treatment for an orthopaedic condition, the procedures can be safely performed simultaneously [167, 168].
2.2.1.7. Hungry Bone Syndrome
A fall in the serum calcium concentration is a common postparathyroidectomy finding in patients with primary, secondary, or tertiary HPT. It usually resolves within 2-4 days. A rapid, severe drop in serum total calcium concentration less than 2.1 mmol/L and/or prolonged hypocalcaemia for more than 4 days postparathyroidectomy is defined as Hungry Bone Syndrome (HBS). The sudden postoperative withdrawal of PTH induces a stop in osteoclastic bone resorption without affecting the osteoblastic activity. Consequently, an increased bone uptake of calcium, phosphate, and magnesium is observed. Risk factors for the development of HBS include preoperative radiographic changes in bone, large parathyroid adenomas, age > 60 years, high numbers of osteoclasts on bone biopsy, and high preoperative levels of serum PTH, calcium, and alkaline phosphatase. Vitamin D-deficient patients are at increased risk of postoperative hypocalcaemia and HBS, which underlines the importance of the preoperative assessment of vitamin D status in all patients with pHPT. Additionally to vitamin D, other factors, such as a high osteocalcin serum level, may be associated with a higher frequency of HBS after parathyroidectomy. Treatment consists of high doses of calcium supplemented by high doses of active metabolites of vitamin D [169-177].
3. CASE REPORT
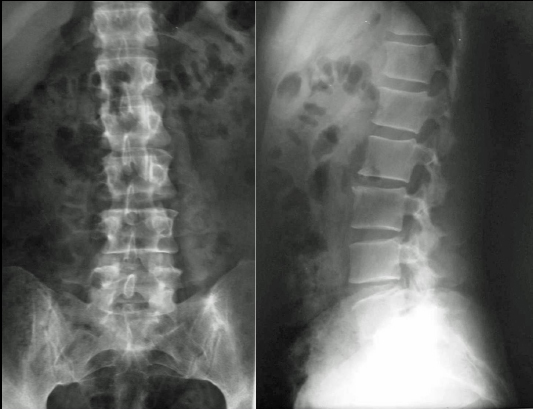
A 47-year-old female entering menopause presented to the outpatient Orthopaedic clinic of our hospital (June, 2001) with a 2-year history (July, 1999) of constant low back pain radiating to her buttocks. The pain was exaggerated when walking or standing but was slightly better when lying in bed. The pain disturbed her during sleep from time to time. A local health care provider had initially diagnosed (April, 2000) mild osteoarthritic changes at the L2–L3 level of the lumbar spine. He reported that on physical examination, there was neither any notable accompanying symptom, such as local tenderness, pattern of radiation, and neurovascular deficit, nor any abnormal findings in the urine test analysis. A potential pathological lumbar fracture, secondary to osteoporosis, was diagnosed 5 months ago after a new private consultation. She additionally complained of lower limb weakness. Plain radiographs by that time (January, 2001), including the chest, lumbar spine, and pelvis, were diagnosed with no abnormal findings (Figs. 1 and 2).
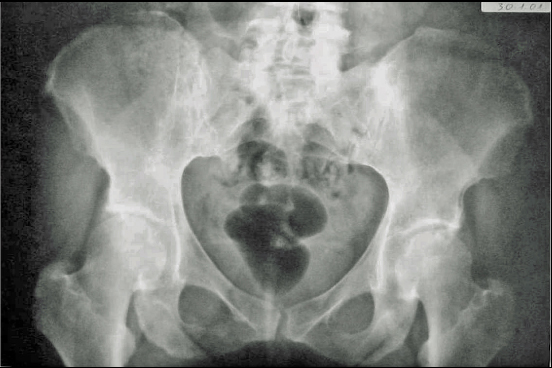
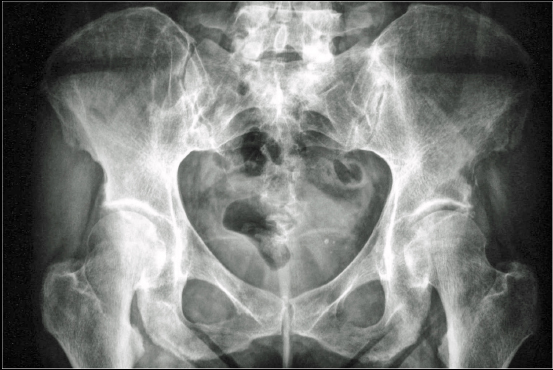
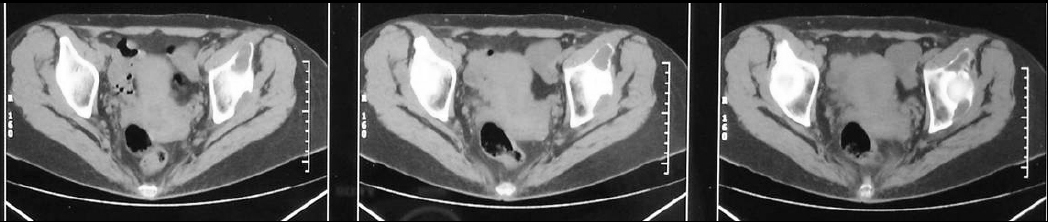
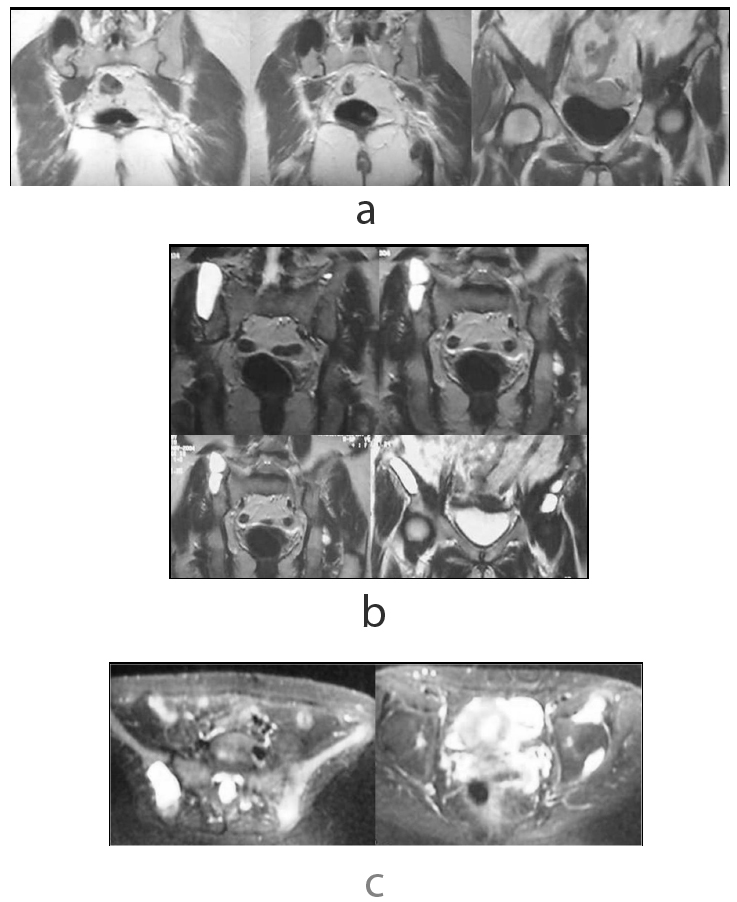
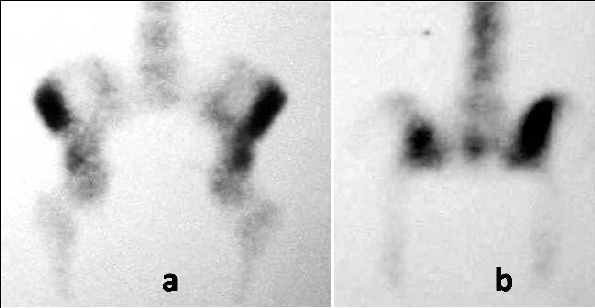
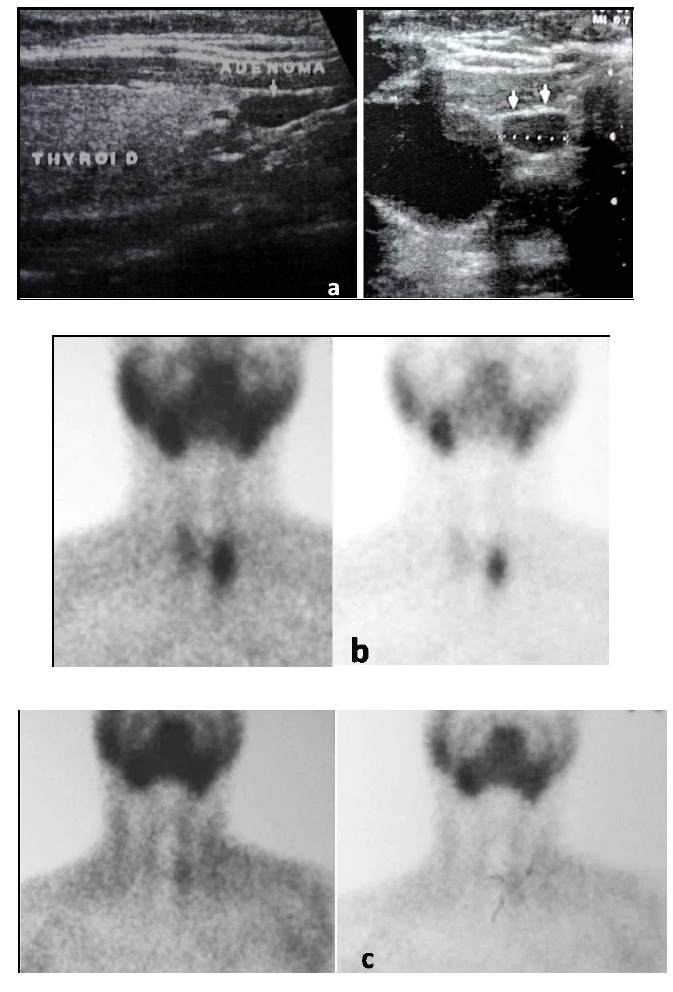
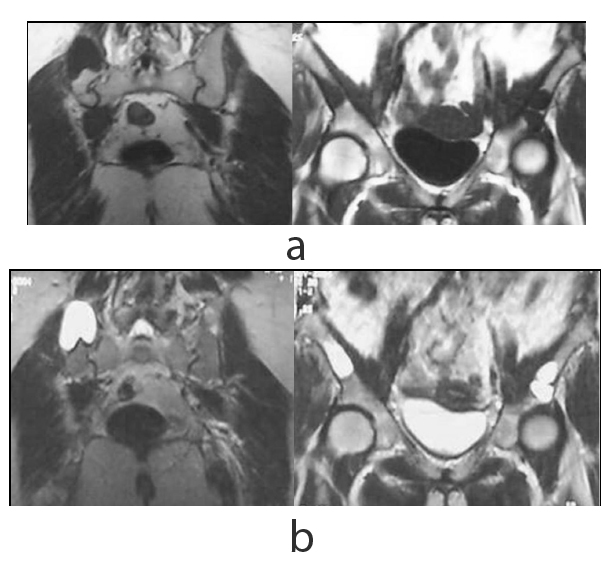
On presentation, the physical examination revealed left hip pain, more pronounced in the last degrees of internal rotation, and right sacroiliac pain. Specific areas of tenderness were localized below the left anterior iliac crest and laterally of the right sacroiliac joint. A new radiograph of the pelvis indicated a lytic lesion above the left acetabulum and another bigger one that was surrounded by a sclerotic rim laterally to the right sacroiliac joint (Fig. 3). A pelvic CT scan revealed that the two well-defined osteolytic lesions affected both the cortical and trabecular microstructure. They were both relatively well demarcated and exhibited bone erosion and cortical attenuation with no evidence of expansion or invading soft tissue (Fig. 4). A subsequent pelvic MRI showed that the lesions were hypointense on T1-weighted images, hyperintense on T2-weighted images, and exhibited the fluid-fluid level appearance, potentially due to intratumoral haemorrhage. The findings were considered consistent with those of an aneurysmal bone cyst, but the appearance of two lesions indicated the potential diagnosis of skeletal or systemic (presenting with typical skeletal and splenic involvement) cystic angiomatosis (Fig. 5). A 99mTc-MDP whole-body bone scan was performed (July, 2001), and it demonstrated elevated uptake in both lesions (Fig. 6). Laboratory studies were then performed (Table 1). The diagnosis of pHPT was established on the basis of the biochemical findings. Imaging studies, including ultrasound and 99mTc sestamibi scans, of the parathyroid glands followed (January, 2002), and they revealed a left inferior parathyroid adenoma (Fig. 7a-c). The patient had a standard parathyroid operation 3 months later (April, 2002), the tumor was removed, and histopathology revealed a parathyroid adenoma. The patient was admitted 4 days postsurgery to the Department of Internal Medicine of our hospital with the diagnosis of postoperative hypoparathyroidism and HBS. The clinical symptoms and signs of hypocalcaemia included numbness and tingling (paraesthesia) in the perioral area as well as in the hands and feet, muscle cramps, and carpopedal spasms. The Trousseau sign of latent tetany, in which the wrist and metacarpophalangeal joints flex, the proximal and distal interphalangeal joints extend, and the fingers adduct, was positive. The sign is also known as ‘main d’accoucheur’ (French for ‘hand of the obstetrician’) because it supposedly resembles the position of an obstetrician’s hand in delivering a baby. There was a paleness of the skin and conjunctivae. Abnormal vaginal bleeding was also diagnosed. Laboratory findings revealed persistent hypocalcaemia. Treatment included blood transfusion, uterine curettage, intravenous calcium infusion, as well as oral administration of calcitriol, calcium supplement, and magnesium. The patient recovered and was discharged from the hospital after 40 days. New pelvic MRI 3 years later (November, 2004) revealed no change in the appearance of the brown tumors (Fig. 8a, b). During the final follow-up (2021), the biochemical parameters, including serum calcium, vitamin D, and PTH, were normal but the pelvic radiograph indicated no significant changes in the appearance of the brown tumors (Fig. 9).
| Laboratory Studies | 20/07/2001 | 02/10/2001 | 11/01/2002 |
|---|---|---|---|
| Haematocrit | 29.4% (36-46) | - | 29% (36-46) |
| Haemoglobin | 9.9 gr% (12-14) | - | 9.5 gr% (12-14) |
| RBC | 3.2 M/μl (4,2-5,4) | - | 3.6 M/μl (4,2-5,4) |
| ESR | 35 mm/1st hour | - | 61 mm/1st hour |
| Total proteins | 8.7 gr% (6-8) | - | - |
| Albumin | 5.6 gr% (4-5.5) | - | - |
| Globulin | 3.1 gr% (2-3) | - | - |
| ALP | 922 U/L (100-290) | 1185 IU/L (80-300) | 1332 U/L (100-290) |
| Calcium | Total 1.94 mgr% (0.98-1.27) | Ionized 2.9 meg/l (2.1-2.7) Total 12.5 mg/dl (8.3-10.4) |
Total 1.93 mgr% (0.98-1.27) |
| 24-hour urine calcium | - | - | 289.5 mg/24h (25-250) |
| PTH | - | - | 1042 pg/ml (10-71) |
| WBC, platelets, Blood sugar, Serum urea, creatinine, phosphate, immunoglobulins, C3, C4, anti-nuclear, anti-DNA, anti-smooth muscle, anti-mitochondrial antibodies, Urine test | Normal values | - | - |

CONCLUSION
A high index of clinical suspicion may be required for the diagnosis of pHPT in a patient presenting in the common orthopaedic practice. A minimal increase of the serum calcium, a mild increase of the ESR, and a significant increase of total ALP were the only initial laboratory findings in the reported case, which had already developed brown tumors of the pelvis.
CONSENT FOR PUBLICATION
Permission to use data and images has been obtained from the patient.
FUNDING
The author received no financial support for this study.
CONFLICT OF INTEREST
The author certifies that he has no commercial associations (such as consultancies, stock ownership, equity interest, patent/licensing arrangements, etc.) that might pose a conflict of interest in connection with the submitted article.
ACKNOWLEDGEMENTS
Declared none.

