All published articles of this journal are available on ScienceDirect.

Treatment of Varus Deformities of the Lower Limbs in Patients with Achondroplasia and Hypochondroplasia
Authors Info & Affiliations
Abstract
Angular deformities of the lower limbs are a common clinical problem encountered in pediatric orthopaedic practices particularly in patients with osteochondrodysplasias. The varus deformity is more common than the valgus deformity in achondroplasia and hypochondroplasia patients because of the unusual growth of the fibulae than that of the tibiae. We retrospectively reviewed six patients (four patients with achondroplasia and two patients with hypochondroplsia) with relevant limb deformities due to the above-mentioned entities. All patients manifested significant varus deformity of the lower limbs. Detailed phenotypic characterization, radiologic and genetic testing was carried out as baseline diagnostic tool. We described the re-alignment procedures, which have been applied accordingly. Therefore, bilateral multi-level procedures, multi-apical planning and limb lengthening have been successfully applied. While recognition of the underlying syndromic association in patients who are manifesting angular deformities is the baseline for proper orthopaedic management, this paper demonstrates how to evaluate and treat these complex patients.
INTRODUCTION
Angular deformities of the lower limbs might accompany a long list of disorders such as osteogenic, myogenic, neurogenic, and endocrinologic ones. The differentiation of these conditions is the baseline in the management. Developmental angular deformities manifest varus angulation centred at the knee (metaphyseal beaking), thickening of the medial tibial cortices, and tilted ankle joints. Congenital angulation might manifests as posteromedial bowing with cortical thickening along the cavity of the curvature and, in some cases, diaphyseal broadening. Angular deformities of the lower limbs are a common clinical deformity in a long list of patients with osteochondrodysplasia such as achondroplasia, and hypochondroplasia [1-4].
Achondroplasia is an autosomal dominant disorder, but approximately 75% of cases represent new dominant mutations. Achondroplasia is caused by mutation in the gene that codes for the fibroblast growth factor receptor type 3 (FGFR3) [5-7]. The abnormality seen in the bone of patients with achondroplasia is failure of enchondral ossification. Intramembraneous and periosteal ossification are undisturbed. Histologic studies have shown disarray of the chondrocytes, with loss of columnisation and loss of normal chondrocyte proliferation. Fibrous tissue is present in the zone of provisional calcification, and those trabeculae which are present, are irregular [8]. Short limbed, short trunked (apparent rhizomelic) dwarfism with relatively long trunk associated with large head with prominent forehead saddle nose, gibbus in thoracolumbar region and bowed legs. Achondroplasia is the most common condition associated with severe disproportionate short stature. The diagnosis can usually be made on the basis of clinical characteristics and very specific features on radiographs, which include contracted base of the skull, square shape of the pelvis with a small sacrosciatic notch, short pedicles of the vertebrae, rhizomelic (proximal) shortening of the long bones, trident hands, a normal-length trunk, proximal femoral radiolucency, and (by midchildhood) a characteristic chevron shape of the distal femoral epiphysis. Other rhizomelic dwarfing disorders such as hypochondroplasia and thanatophoric dysplasia are part of the differential diagnosis, but achondroplasia usually can be distinguished from them because the changes in hypochondroplasia are milder and the changes in thanatophoric dysplasia are much more severe and invariably lethal.
Hypochondroplasia is an autosomal dominant disorder, characterized by mild, markedly variable short-limb dwarfism and a thick body build. Macrocephaly, frontal bossing, broad and stubby hands and feet as well as mild generalized joint laxity are frequent abnormalities. Long bones are short, with wide-appearing diaphysis, mild flaring of metaphyseal-epiphyseal junction, slight shortening of ulna relative to radius, elongated ulnar styloid, elongated distal fibula, short and broad femoral neck and rectangular proximal tibial epiphyses are frequent abnormalities. Hypochondroplasia is caused by mutation in the gene that codes for the FGFR3 [5-7]. Hypochondroplasia cannot be detected at birth and, if mild, may remain undiagnosed throughout the patient’s life. Clinically, patients with hypochondroplasia are short, but less so than those with achondroplasia. The spectrum of severity is wide, ranging from severe short-limbed dwarfism to short, apparently normal prepubertal children who manifest disproportion only after failure to achieve a pubertal growth spurt. The ratio of sitting height to standing height is increased, but the body disproportion may not be apparent until puberty. Final height has been reported between 118-165 cm. The head appears only slightly enlarged compared with the limbs, but the forehead is large and high. Some patients have small hands. Short stature is one of the parental concerns. Pre-operative anteroposterior lower limbs radiographs in two patients with hypochondroplasia showed elongated distal fibulae, short and broad femoral neck and rectangular proximal tibial epiphyses were apparent (Fig. 2). Growth hormone therapy remains investigational, though the response to growth hormone is greatest during the first year of use. Though, the response varies among patients, perhaps because of the genetic heterogeneity of the disease, although it is known that patients with hypochondroplasia respond more favourably to growth hormone therapy with greater increase in height than those with achondroplasia [9].
The purpose of this study is to present our experience in the treatment of the above-mentioned entities, and to discuss common treatment algorithms in the literature for management of dysplasia-associated lower limb angular deformities.
PATIENTS AND METHODS
This study was approved by the local ethics committee (Medical University of Vienna, EK Nr. 921/2009), and informed consents were obtained from the patients guardians.
We underwent a retrospective chart review of six patients with different ethnic origins (four patients with achondroplasia and two patients with hypochondroplasia) with respect to prominent skeletal disorders and subsequent treatment course. These patients’ records were reviewed in the Osteogenetic Department of the Orthopaedic Hospital of Speising, Vienna, Austria.
Detailed phenotypic and genotypic characterization has been performed in all our patients. First, all patients with achondroplasia and hypochondroplsia have been genetically tested and showed FGFR3 mutations. Complete general and neurologically oriented physical examination was given. Continuous assessment of history and MRI imaging’s for possible obstructive sleep apnea and prompt awareness was given to serious symptomatology such as regarding glottal stops, chocking, intermittent breathing, apnea, deep compensatory sighs, secondary enuresis, and recurrent night-time awakening or emesis.
Varus deformity was the primary orthopaedic abnormality in achondroplastic and hypochondroplastic patients. The following steps have been carefully considered for our achondroplastic and hypochondroplastic patients who underwent surgical interventions with anaethesia; because of the narrow foramen magnum and to avoid unintentional spinal cord compression, special care has been made in manipulation the neck [10]. We usually avoid spinal anaesthesia especially in children with severe lumbar lordosis, because of limited space within the spinal canal [11].
Achondroplastic Patients
AP radiographs of the lower limbs in three boys and one girl of 6, 7, 8 and 9 years-age, respectively, were assessed. All manifested the characteristic radiographic features of achondroplasia: shortness of the tubular long bones, with a relative increase in bony diameters and densities were apparent. The metaphyses were widened and flared, but the epiphyses were uninvolved. The growth plates were U-shaped or V-shaped. This was best seen at the distal femur. The long bones, especially the tibiae, were bowed. The pelvis characteristically appeared broad and flat, with squared iliac wings. The ilium appeared broad because the pelvis was formed almost entirely by intramemberanous ossification, which was undisturbed in achondroplasia. The sciatic notches were small and the acetabuli were horizontal. Genu vara was a constant skeletal deformity encountered. In addition there was a relative shortening of the tibia compared to the fibular length. This tibial shortening was typically associated with a relevant ankle joint varus evolving during growth. The varus deformity ranged between 15 to 20°, and genu varum was the most common deformity (Fig. 1a-d).
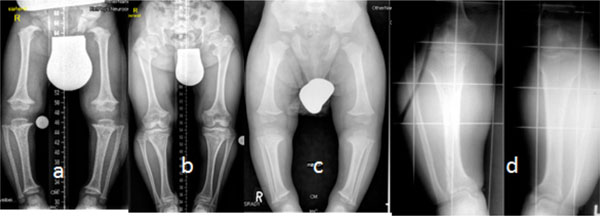
(a-d) AP radiographs of the lower limbs in achondroplasia (three boys and one girl of 6, 7, 8 and 9 years-old respectively), all manifested the characteristic radiographic features of achondroplasia; shortness of the tubular long bones, with a relative increase in bony diameters and densities are apparent. The metaphyses are widened and flared, but the epiphyses are uninvolved. The growth plates are U-shaped or V-shaped. This is best seen at the distal femur. The long bones, especially the tibiae, are bowed. The pelvis characteristically appears broad and flat, with squared iliac wings. The ilium appeared broad because the pelvis is formed almost entirely by intramemberanous ossification, which is undisturbed in achondroplasia. The sciatic notches are small and the acetabuli are horizontal. Genu vara was a constant skeletal deformity encountered. In addition there was a relative shortening of the tibia compared to the fibular length. This tibial shortening is typically associated with a relevant ankle joint varus evolving during growth, the varus deformity ranged between 15 to 20°.
SURGICAL TECHNIQUES AND RESULTS FOR ACHONDROPLASTIC PATIENTS
Multi-apical planning has been performed due to the varus alignment of the bone, which was caused by both the proximal tibia (reduced mMPTA) and the distal tibia (increased mLDTA).
A multi-apical planning for correction of the deformity was considered. The bi-level osteotomies were performed on the intersection between the proximal and distal mechanical axis lines, respectively, and a third line drawn as representative as possible of the mid-diaphysis (Fig. 2). Continuous distraction osteogenesis in a 9-year-old girl has already resulted in a respectable total gain of length and restoration of tibial alignment (Fig. 3). Similar correction in a-7-year-old achondroplastic boy showed correction of the deformity by means of bilevel procedure (Fig. 4). The final results of four children with achondroplasia after removal of the lengthening device showed straight, re-aligned legs. Thus, feet are notably better positioned than prior to the correction, d) long-leg standing radiograph of the frontal plane mechanical axis showed optimal limb alignment (Fig. 5). There were no complications noted.

Multi-apical planning has been performed due to the varus alignment of the bone, which is caused by both the proximal tibia (reduced mMPTA) and the distal tibia (increased mLDTA), a multi-apical planning for correction of the deformity is considered. The bilevel osteotomies are performed on the intersection between the proximal and distal mechanical axis lines, respectively, and a third line drawn as representative as possible of the mid-diaphysis.

AP radiograph are showing the correction of the deformity in a 9-year-old achondroplastic girl by means of the bilevel procedure. Continuous distraction osteogenesis has already resulted in a respectable total gain of length and restoration of tibial alignment. However, the bone regenerate has by far not been consolidated, showing a ventral deficit in the lateral view radiograph.

AP radiograph are showing the correction of the deformity in an 7-year-old achondroplastic boy, by means of the bilevel procedure. Continuous distraction osteogenesis has already resulted in a respectable total gain of length and restoration of tibial alignment.
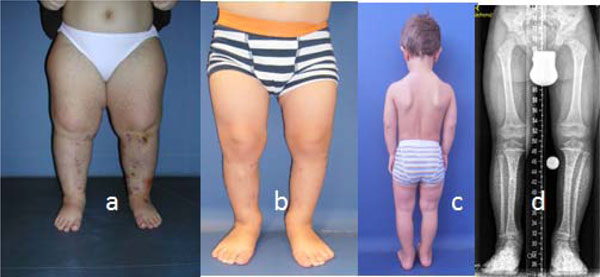
(a-c) The final results of four children with achondroplasia after removal of the lengthening device showed straight, re-aligned legs. Thus, feet are notably better positioned than prior to the correction, (d) long-leg standing radiograph of the frontal plane mechanical axis showed optimal limb alignment.
Hypochondroplastic Patients
AP radiographs in a-7 and 10-years-old- patients with hypochondroplasia revealed both an increased distal femoral and an increased proximal tibia varus, respectively, whereas the distal tibia was almost normally configured. In contrast to achondroplasia the femoral condyle and tibial plateau was much less widened and flared (Fig. 6a, b).

(a, b) AP radiographs of a-7 and 10 year old boys with hypochondroplasia with variable degrees of genu varum. Long bones are short, with wide-appearing diaphysis, mild flaring of epiphyseal-metaphyseal junction. Elongated distal fibulae, short and broad femoral neck and rectangular proximal tibial epiphyses were apparent. There was an increased distal femoral and an increased proximal tibia varus, whereas the distal tibia is almost normally configured. In contrast to achondroplasia the femoral condyle and tibial plateau is much less widened and flared.
SURGICAL TECHNIQUES AND RESULTS FOR HYPOCHONDROPLASTIC PATIENTS
Photo showed the mounted bilateral TSF in a 16-year-old boy. After the lengthening procedure, the tibial bowing was sufficiently corrected. In this case, a single-level osteotomy was indicated to restore alignment (Fig. 7). An additional procedure was necessary to correct the femoral deformity, located at the distal part of the bone. A solid pin fixation of the device fairly proximal and distal to the osteotomy site was tremendously important for stability during the lengthening procedure (Fig. 8). Long-leg standing radiograph six years after femoral and nine years after tibial lengthening: The frontal plane mechanical axis, which was drawn from the center of the femoral head to the center of the ankle plafond, passed slightly medial to the center of the knee joint indicating an optimal limb alignment. This perpendicular distance, also called mechanical axis deviation (MAD), is normally 8±7mm medial to the center of the knee joint (Fig. 9). There were also no complications noted.
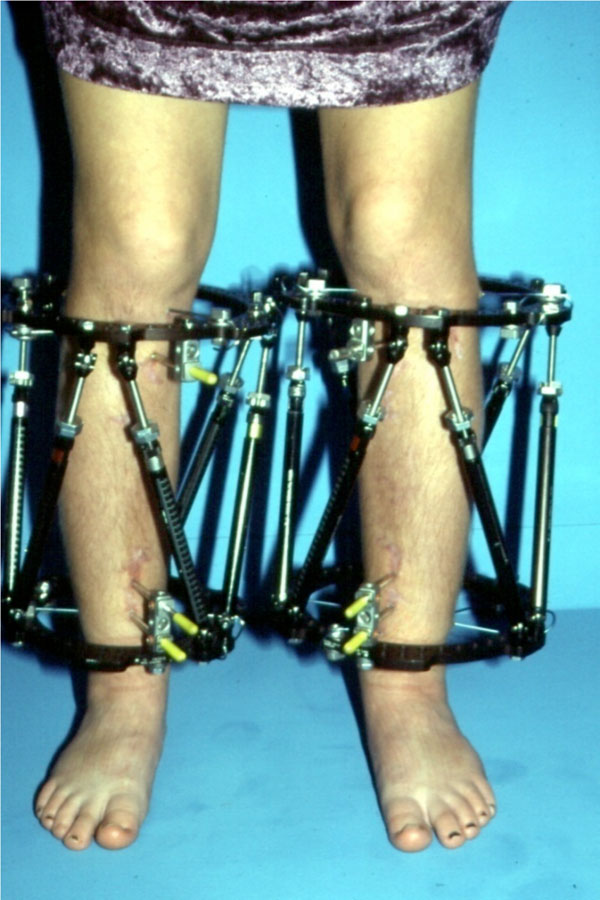
Photograph of a-16-year-old boy with hypochondroplasia showed the mounted bilateral TSF. After the legthening procedure, the tibial bowing was sufficiently corrected. In this case, a singlelevel osteotomy was indicated to restore alignment.

An additional procedure was necessary to correct the femoral deformity, located at the distal part of the bone. A solid pin fixation of the device fairly proximal and distal to the osteotomy site is tremendously important for stability during the lengthening procedure.
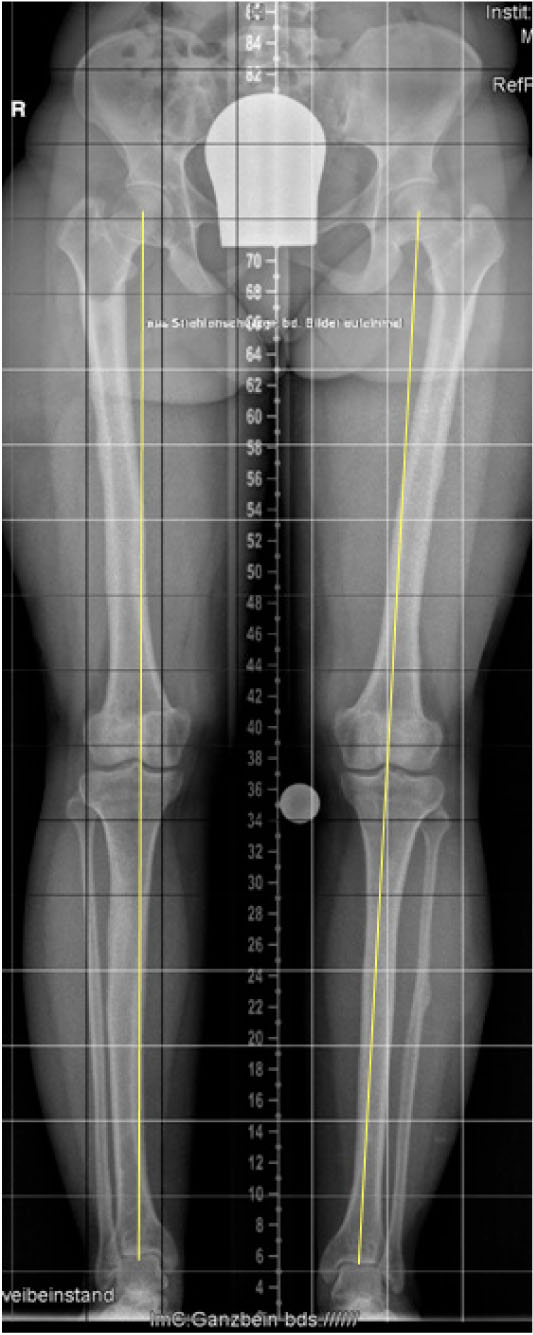
Long-leg standing radiograph six years after femoral and nine years after tibial lengthening: The frontal plane mechanical axis, which is drawn from the center of the femoral head to the center of the ankle plafond, passes slightly medial to the center of the knee joint indicating an optimal limb alignment. This perpendicular distance, also called mechanical axis deviation (MAD), is normally 8±7mm medial to the center of the knee joint.
DISCUSSION
Angular deformities of the tibia and fibula are probably one of the most common and obvious abnormalities that affect a child’s lower extremity encountered in orthopaedic departments. They can be congenital or acquired, physiologic or pathologic, but all, because of the real or apparent detrimental effect that they have on gait and upright activity draw immediate attention to themselves. The majority of lower leg deformities are in fact physiologic and resolve spontaneously, thus early recognition of the benign nature of such deformities is an important as the correct diagnosis of true pathologic conditions is mandatory.
Achondroplasia is the most common condition associated with disproportionate short stature. Substantial information is available concerning the natural history and anticipatory health supervision needs in children with this dwarfing disorder. Most children with achondroplasia have delayed motor milestones, problems with persistent or recurrent middle-ear dysfunction, and bowing of the lower legs.
Surgical treatment to re-align the lower limbs in achondroplasia is generally indicated for cases who present either a severe, cosmetically unacceptable or clinically symptomatic limb deformity. Varus malalignment is generally more common than genu valga in this syndrome. Realignment can generally be achieved by gradual correction using external fixation devices. Other methods such as fibula epiphysiodesis have in contrast shown to be of controversial value. Limb lengthening is usually performed during adolescence and aims to increase height in one or more lengthening steps of about 5-8cm each. Due to modern external fixation devices (i.e. TSF) lengthening can easily be combined with correction of frontal malalignment in achondroplasia patients. Further skeletal corrections (i.e. of thoracolumbar kyphosis, spinal stenosis) should be indicated depending on the severity of the case and symptoms [2, 4, 11].
Hypochondroplasia in contrast is a mild or incomplete form of achondroplasia, and often underdiagnosed (one of the most frequent autosomal dominant disorders [9]). Patients are less severely affected than in achondroplasia, and the two conditions breed true. The diagnosis might be difficult in the neonatal period, despite the presence of mild rhizomelic limb shortening and some bossing of the forehead. It has been said in the past that patients with hypochondroplasia do not have frontal prominence, but this is not always the case. Patients with hypochondroplasia can similarly be treated by limb lengthening to achieve an almost normal age-specific body height. In this syndrome, the same devices can be used as mentioned above for achondroplasia. However, special attention should be paid to any associated varus or valgus deformities, which should be addressed accordingly [8, 11].
As the molecular pathways involved in the pathogenesis of achondroplasia and hypochondroplsia become clearer, a number of potential therapeutic strategies have emerged.
Selective FGFR3 kinase inhibitors have been developed and show promise in cell and organ culture experiments, but to date none have worked effectively in whole animals. An alternative approach has involved generating antibodies to block FGFR3 activation. Highly specific humanized antibodies have been developed. Although these antibodies block receptor activation in cell culture, in-vivo studies have yet to be done. The therapeutic use of CNP or a CNP analog that could activate the NPR-B signaling pathway to counter excessive FGFR3 signals has been proposed. This approach is appealing because other natriuretic peptides have been used clinically for their hemodynamic effects in adults and even in children. Although they seem to be safe, at least in the short term, a major drawback is their very short half-life, which requires them to be administered by continuous infusion. A variation of this approach involves therapeutically targeting NPR-C, another natriuretic peptide receptor that binds to CNP. NPR-C, which is present on hypertrophic chondrocytes in the growth plate, lacks the ability to increase intracellular cGMP and has been proposed to function as a clearance receptor to down-regulate the effects of natriuretic peptides. Theoretically, blocking NPR-C would lead to an increase in CNP available to bind to NPR-B, which would be expected to antagonise FGFR3 signals in the growth plate [6, 12].
There are two considerations with regard to molecular treatment of achondroplasia and hypochondroplasia that deserve special attention. The first is that treatment would need to be long term, probably starting soon after birth when the diagnosis is made and lasting through puberty. Because skeletal size is usually only mildly reduced at birth, there would potentially be ample time for catch-up growth. However, this long period of treatment adds challenges to any therapeutic approach. The second consideration involves targeting therapeutic agents to the cartilaginous growth plate. Compared with most tissues, cartilage is avascular and the dense and highly charged extracellular matrix that surrounds chondrocytes represents a formidable barrier for drug delivery. Indeed, these factors might explain, at least in part, why treatments that have worked in cell and organ culture experiments have failed in whole animals. Agents given systemically might need to be administered in higher doses than those used for most other tissues to achieve therapeutic levels in the growth plate, and this could lead to side-effects in the other tissues [13, 14].
SUMMARY
Osteochondrodysplasias are a heterogeneous group of genetic skeletal dysplasias. Patients with osteochondrodysplasia are considered chronic clients in most of the pediatric departments because of diverse skeletal deformities. The phenotypic characterization and whenever possible the genotypic correlation are the baselines for proper management. The role of the physician to determine if the deformity is physiologic or in connection with an intrinsic bone disorder or other aetiological background is fundamental.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
We wish to thank Miss Rima Al Kaissi, University of Vienna for her help in providing the required literature.

