All published articles of this journal are available on ScienceDirect.

Cryopreservation Effect on Proliferative and Chondrogenic Potential of Human Chondrocytes Isolated from Superficial and Deep Cartilage
Authors Info & Affiliations
Abstract
Objectives:
To compare the proliferative and chondrogenic potential of fresh and frozen chondrocytes isolated from superficial and deep articular cartilage biopsies.
Materials and Methodology:
The study included 12 samples of fresh and frozen healthy human knee articular cartilage. Cell proliferation was tested at 3, 6 and 9 days. Studies of mRNA quantification, protein expression and immunofluorescence for proliferation and chondrogenic markers were performed.
Results:
Stimulation of fresh and frozen chondrocytes from both superficial and deep cartilage with fetal bovine serum produced an increase in the proliferative capacity compared to the non-stimulated control group. In the stimulated fresh cells group, the proliferative capacity of cells from the deep biopsy was greater than that from cells from the superficial biopsy (0.046 vs 0.028, respectively, p<0.05). There was also a significant difference between the proliferative capacity of superficial zone fresh (0.028) and frozen (0.051) chondrocytes (p<0.05). CCND1 mRNA and protein expression levels, and immunopositivity for Ki67 revealed a higher proliferative capacity for fresh articular chondrocytes from deep cartilage. Regarding the chondrogenic potential, stimulated fresh cells showed higher SOX9 and Col II expression in chondrocytes from deep than from superficial zone (p<0.05, T student test).
Conclusions:
The highest rate of cell proliferation and chondrogenic potential of fresh chondrocytes was found in cells obtained from deep cartilage biopsies, whereas there were no statistically significant differences in proliferative and chondrogenic capacity between biopsy origins with frozen chondrocytes. These results indicate that both origin and cryopreservation affect the proliferative and chondrogenic potential of chondrocytes.
INTRODUCTION
The limited ability of articular cartilage to repair arises from its avascular nature [1-3]. Consequently, progenitor cells in blood and marrow cannot enter a damaged region to influence or contribute to the repair process [4]. There are three categories of joint damage according to their characteristics: 1) minor damage or repeated trauma of the matrix and cells, 2) surface damage or chondral fracture or damage that do not penetrate the subchondral bone, and 3) osteochondral damage that penetrate the subchondral bone [5]. Reliable techniques and methods for stimulating growth of new cartilage tissue to treat degenerative diseases and trauma are lacking [6]. At present, most techniques for repairing injuries of articular cartilage aim to overcome its limitations for healing injuries by facilitating access of the vascular system to the injury [2]. However, these techniques generate a fibrocartilaginous repair tissue that lacks both the structure and function of natural hyaline articular cartilage. To avoid the need for prosthetic replacement, cell treatments have been developed that attempt to generate repair tissue with the structure, biochemical composition and function similar to natural articular cartilage [7].
Cell therapy using autologous chondrocyte implantation (ACI) to repair focal defects of articular cartilage, obtains a repair tissue of higher quality compared to fibrocartilage tissue [7-16]. This therapeutic procedure is used clinically in combination with a periostial cover to treat chondral or osteochondral defects of the knee, with good clinical results [4]. ACI involves obtaining articular cartilage explants by arthroscopy from areas not under load. Chondrocytes isolated from these cartilage biopsies are cultured in the laboratory under in vitro conditions until an adequate number of cells for implantation into the chondral defect is obtained [17]. However, ACI has some limitations [18-27], such as basal and peripheral integration, inadequate fill, fibrous, rather than hyaline repair, etc. Moreover, the proliferation and chondrogenic potential of chondrocytes is also an important limitation in the ACI practise.
The senescence of human articular chondrocytes reduces their proliferative capacity in vitro. Proliferating cell nuclear antigen (PCNA), cyclin D1 (CCND1) and Ki67 are molecular markers with predictive capacity for cell proliferation. PCNA is expressed exclusively in actively proliferating cells but not in non-cycling cells [28]. CCND1 belongs to a regulatory proteins family that drive the ordered progression of mammalian cells through critical transition points in the cell division cycle. The D-type cyclins control the passage of cells through the G1 phase, allowing entry into the S phase. Furthermore, assessment of cell proliferation by detecting the Ki67 antigen in cell populations has been used to determine a wider range of dividing cells. Ki67 antigen is co-expressed during late G1, S, G2, and M phases, allowing for a wider range of proliferating cell populations than cyclin D1 [29]. But success cell therapy or tissue engineering do not reside just in the proliferative capacity, chondrogenic potential is also critical. Col II (type II collagen), that encodes a key cartilage-specific extracellular matrix protein [30], and SOX9 (SRY sex determining region-Y-box 9), that encodes a nuclear transcription factor [31], are two molecular markers predictive of the chondrogenic potential of human articular chondrocytes cultured in monolayer. SOX9 is required for the deposition of cartilage matrix containing collagens II, IX, XI and aggrecan [32]. This transcription factor is expressed continuously in chondrocytes up to the hypertrophic stage [33]. In this regard, the main objective of this study was to compare the proliferative and chondrogenic potential of fresh and frozen chondrocytes isolated from superficial and deep articular cartilage biopsies.
MATERIALS AND METHODOLOGY
Sample Processing and Cell Culture Techniques
Collection and Obtaining of Articular Cartilage Samples
The Ethic Committee of Clinical Investigation of Galicia approved this study. Samples of knee articular cartilage were provided by the Autopsy Service at CHU A Coruña (Spain). Cartilage sections (N=12) were dissected from the femoral condyles and tibial plate of each knee articular cartilage under sterile conditions and washed with Dulbecco's Modified Eagle's Medium (DMEM) (Gibco, Life Technologies, Paisley, Scotland) supplemented with penicillin (100 units/ml) and streptomycin (100 μg/ml) (Gibco Invitrogen, Grand Island, NY, USA). From each specimen, superficial and deep cartilage samples were separated, minced with a scalpel and transferred to a digestion buffer containing 1% trypsin (Sigma-Aldrich, St. Louis, MO, USA) for 15 min at 37ºC. Superficial cartilage comprised 0.5 mm thickness and was obtained in a single layer from one of the condyles, the remaining cartilage was considered as deep cartilage. Therefore, deep cartilage comprised a mixture of chondrocytes from the middle and deep zones of cartilage. Regarding the superficial and deep cartilage, around 100,000 cells were successfully isolated per gram of tissue with a 90% viability. All superficial and deep cartilage biopsies were analyzed by hematoxylin-eosin staining in order to determine the correct selection of the layers. Following removal of trypsin, the cartilage samples were incubated for 12 to 16 h in a digestion solution containing 5 CDU (collagen digestion units) type IV collagenase (Sigma-Aldrich, St Louis, MO). After this digestion solution was removed, the cells were washed with DMEM and centrifuged at 200xg for 10 minutes. The number of isolated chondrocytes was counted using a Neubauer Chamber and 0.4% trypan blue dye (Sigma-Aldrich, St Louis, MO, USA) to assess viability. Four aliquots with equal numbers of cells were separated, two each from superficial and deep cartilage (Fig. 1). One aliquot of cells from each superficial and deep cartilage sample was cultured in DMEM with 10% fetal bovine serum (FBS) to perform cell proliferation assays. The other two aliquots of cells from superficial and deep cartilage samples were cryopreserved at -196°C for 3 months following the controlled freezing program. For controlled freezing we used freezing rates of 1ºC/min to a temperature of -40ºC, 2ºC/min to -60ºC, and 5ºC/min to -150ºC. The equipment used for controlled freezing was CM 25 (Carburos Metálicos, Barcelona, Spain). For thawing protocol we introduced the freeze aliquot on a heat-bath at 37ºC. After thawing, cells were introduced in a tube with DMEM plus 10% FBS and centrifuged at 200xg for 10 min. These chondrocytes were cultured, then, following two sub-cultures, the same proliferation studies described above for fresh chondrocytes were performed.
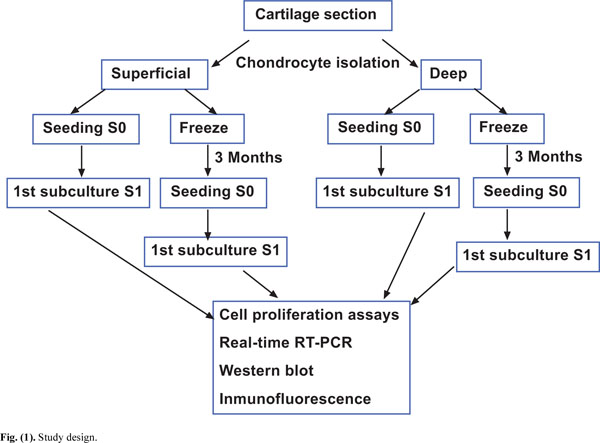
Study design.
Chondrocyte Culture
Chondrocytes were cultured in 25 cm2 culture flasks with DMEM supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, 1% glutamine and 10% FBS (Gibco, Life Technologies) in a humidified 5% CO2 atmosphere at 37ºC (Steri-Cult 200 Incubator HEPA class 100, Hucoa Erlöss, Madrid, Spain). Two weeks later, when confluency was attained, the cells were sub-cultured and used for proliferation studies.
Cell Proliferation Assays
Cells, grown on DMEM with 10% FBS at passage 1, from both superficial and deep cartilage samples were placed in 12-well plates (Corning Incorporated, Corning, NY, USA) at 5x104 cells per well (1ml per well) and cultured in DMEM medium without FBS for 24 hours. The culture medium was then removed and the cells were stimulated by culturing with DMEM with 10% FBS (Sigma-Aldrich, St. Louis, MO, USA). For the control cultures, DMEM without FBS was used. At 3, 6 and 9 days after stimulation, cells were washed with 1% phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde for 15 minutes. Following paraformaldehyde removal, two washes with 1% PBS were performed before adding 0.5% toluidine blue for 15 minutes. The toluidine blue was then removed and the cells were again washed with 1% PBS to remove unbound dye. The cells were solubilized with 1.5 ml of 1% SDS and the absorbance was read on a spectrophotometer at 570 nm. The results shown in the graphs represent the differences in absorbance obtained for samples cultured in DMEM with 10% FBS relative to the control culture in DMEM without FBS at each time point. Each value represents the average of two independent experiments.
Molecular Biology Techniques
RNA Extraction
Isolation of total RNA from cultured cells was performed using Trizol Reagent (Invitrogen, Madrid, Spain), following the manufacturer´s protocol. From each cell culture, 106 cells were obtained for RNA isolation. Total RNA was further processed by reverse transcriptase-polymerase chain reaction (RT-PCR) or stored at -80ºC until used. RNA integrity was confirmed using 2% agarose gel electrophoresis stained with ethidium bromide. RNA also was assessed for quantity at 260 nm using a NanoDropTM spectrophotometer (Fisher Scientific, Madrid, Spain). The A260/A280 ratio was calculated for quality and purity.
cDNA Synthesis
RT-PCR was performed from 1 µg of total RNA using SuperScriptTM First-Strand Synthesis System for RT-PCR (Invitrogen, Madrid, Spain) to a total volume of 20 µl in a Thermocycler (Gene Amp PCR System 9700, Applied Biosystem, Madrid, Spain). 1 µg of total RNA, 2.5 nM random hexamers, 0.5 mM of dNTP mix, and 3 µl of DEPC-treated water were denatured at 65ºC for 5 minutes and chilled on ice for at least 1 minute. 2 µl of 10xRT buffer, 5 mM MgCl2, 0.01 M DTT, and 40 U of RNaseOUT Recombinant Ribonuclease Inhibitor were mixed, collected by centrifugation, and incubated at 25ºC for 2 minutes. After incubation, 50 U of SuperScriptTM RT were added and incubated at 25ºC for 10 minutes, 42ºC for 50 minutes and 70ºC for 15 minutes in a Thermocycler. Finally, samples were chilled on ice and incubated with 2 U of RNAse H for 20 minutes at 37ºC before proceeding to amplification of the target cDNA. Samples were stored at -20ºC before the cDNA target was amplified. Positive and negative controls were included in each experiment. RNA extraction, reverse transcription-PCR assay setup and post-reverse transcription-PCR product analysis were carried out in separate dedicated rooms to prevent cross-contamination.
Quantitative Real-Time Reverse Transcriptase-PCR Analysis (qRT-PCR)
qRT-PCR analysis was performed using the primers and conditions shown in Table 1. A LightCycler® 480 Instrument (Roche, Mannheim, Germany) using the LightCycler 480 SYBR Green I Master (Roche, Mannheim, Germany) was employed. The PCR reaction consisted of 10 µl of Master Mix 2x conc., 0.35 µM of each forward and reverse primer, the cDNA template (obtained by reverse-transcripton from 1µg RNA) and PCR-grade water up to a final volume of 20 µl in the LightCycler 480 Multiwell Plate 96. This multiwell plate was centrifuged at 1500xg for 2 minutes and was loaded in the LightCycler 480 Instrument until the PCR program started. An initial activation at 95ºC for 5 minutes was followed by an amplification target sequence of 50 cycles at 95ºC for 10s, 60ºC for 10s, and 72ºC for 7s. For melting curve analysis, 1 cycle at 95ºC for 5s, 70ºC for 15s, and 95ºC for 1s was used. The final step was cooling at 40ºC for 10s. We verified that the amplifications and the expected size of each PCR product were specific. 1.8% agarose gel electrophoresis of all PCR products revealed a single band that corresponded to the single-amplified products as predicted by the melting curve analysis of the PCR. PCR primers for mRNA amplification were carefully designed using the web-based ProbeFinder software (Universal ProbeLibrary Design Center) accessible at [34] or via Roche Applied Science home page [35]. PCR primers have been positioned to span exon-intron boundaries, reducing the risk of detecting genomic DNA. Primers were purchased from Roche (Mannheim, Germany). The selection of a suitable housekeeping gene was made using the Human Endogenous Control Gene Panel (TATAA Biocenter, Göteborg, Sweden). The Excel macro named GeNorm VBA applet for Microsoft Excel available at [36] was used to determine the gene(s) with most correlated expression in the set of samples. The selected housekeeping gene, HPRT (hypoxanthine-guanine phosphoribosyltrans-ferase), was used as internal control as well as to verify the integrity of the RNA and efficacy of reverse transcription. Any specimen with inadequate housekeeping HPRT mRNA was excluded from the study. Data analysis was performed using LightCycler 480 Relative Quantification software (Roche, Mannheim, Germany). Relative levels of expression were calculated by the 2-ΔΔCt method of [37]. Each assay was done at least in triplicate and included marker-positive and marker-negative controls and reagent with no template controls. Data were normalized against the lowest value obtained for each gene, which was assigned the value of 1, and were measured as relative expression levels (R.E.L).
Primer Sequences, Conditions and Annealing Temperature of qRT-PCR Assays
| Primersf | Sequence | Length | %GC | Annealing Temperature | Amplicon (bp) | Reference |
|---|---|---|---|---|---|---|
| CCND1 1F | 5´ gac ctt cgt tgc cct ctg t 3´ | 19 mer | 58 | 60°C | 101 | Universal ProbeLibrary |
| CCND 1R | 5´ ggt tca ggc ctt gca ctg 3´ | 18 mer | 61 | |||
| SOX9 2F | 5´gtacccgcacttgcacaac 3´ | 19 | 58 | 60°C | 72 | Universal ProbeLibrary |
| SOX9 2R | 5´ tcgctctcgttcagaagtctc 3´ | 21 | 52 | |||
| COL II 3F | 5´ gtgtcagggccaggatgt 3´ | 18 | 61 | 60°C | 116 | Universal ProbeLibrary |
| COL II 3R | 5´ tcccagtgtcacagacacagat 3´ | 22 | 50 | |||
| HPRT 1F | 5´ tga cct tga ttt att ttg cat acc 3´ | 24 mer | 33 | 60°C | 102 | Universal ProbeLibrary |
| HPRT 1R | 5´ cgagcaagacgttcagtcct 3´ | 20 mer | 55 |
CCND1 = cyclin D1; SOX9 = SRY (sex determining region-Y)-box 9; COL II = type II collagen; HPRT = Hypoxanthine-guanine phosphoribosyltransferase.
DNA Sequencing Analysis
At least one PCR product coming from each qRT-PCR experiment was used as DNA template in order to verify the specificity of the amplified amplicon. PCR products were purified by an enzymatic method (ExoSAP-IT, Amersham Biosciences, Madrid, Spain). DNA sequencing was performed in a reference facility on ABI 3100 (Applied Biosystems, Madrid, Spain) using Big Dye terminators. Forward and reverse specific primers were used (these primers were the same as for the qRT-PCR experiments, see Table 1).
Standard procedures for manipulation of nucleic acids were essentially those of [38].
Western Blot
Cultured cells were washed in ice cold PBS, pH 7.5, and lysed in 0.2 M Tris-HCl, pH 6.8, containing 2% SDS (sodium dodecyl sulphate), 20% glycerol, 1 μg/ml inhibitor cocktail, and 1 mM PMSF (phenyl methyl sulphonyl fluoride (Sigma-Aldrich, St Louis, MO, USA) buffer. Samples were boiled at 100ºC for 5 minutes. Protein concentrations were measured using a NanoDropTM spectrophotometer (Fisher Scientific, Madrid, Spain). 30 µg of protein extract per well was resolved on 10% SDS-polyacrylamide gel and transferred to polyvinylidene difluoride membranes (Immobilon P, Millipore Co, Bedford, MA, USA). The membranes were blocked in 10 mM Tris-HCl pH 7.5, containing 100 mM NaCl, 1% Tween 20, and 5% non-fat dried milk for 1 hour at room temperature (RT) and then incubated overnight with mouse anti-human Cyclin D1 (BD Biosciences, Madrid, Spain) in fresh blocking solution at 4ºC. After several washes, CCND1 protein was detected by incubation with a secondary antibody, ECL anti-mouse IgG horseradish peroxidase-linked whole antibody (GE Healthcare Life Science, Buckinghamshire, UK). To confirm that equal amounts of total proteins were charged, each membrane was incubated with anti-α-tubulin (Sigma-Aldrich Quimica, Madrid, Spain). Data were normalized against the lowest value obtained for CCND1 protein expression levels, assigned the value of 1, and were measured as R.E.L.
Immunofluorescence and Immunohistochemical Techniques
Frozen and deparaffinized and hydrated paraffin sections (4 μm thick) were incubated with anti-human Ki67 antibody (Novocastra, Newcastle, UK). For immunofluorescence analysis, goat anti-mouse-retinal pigment epithelium (RPE) (Dako Diagnósticos, Barcelona, Spain) was used as a secondary antibody. Cells were counterstained with the nuclear fluorophore, DAPI (4 ', 6-diamidino-2-phenylindole, dihydrochloride) (Invitrogen, Madrid, Spain). For immunohistochemical studies, the Kit EnvisionTM Detection Systems Peroxidase/DAB Rabbit/Mouse (Dako Diagnósticos, Barcelona, Spain) was used to determine antigen-antibody interactions. Negative controls were created by omitting the primary antibody.
Quantification of the cells positive for Ki67 was performed using analiSIS® software (version D; Olympus, Barcelona, Germany). The percentage of positive cells for Ki67 was assessed counting the same number of cells in both cartilage zones, comparing sample to sample, and always respecting the same area of analysis of the samples. Quantification using the software must be coincident with the microscopic screening. The nuclear size of positive cells was taken into account, i.e. there was no greater percentage of positivity as a greater nuclear size, since differences between cells have been respected.
Statistical Analysis
All statistical analyses were performed using SPSS 16.0 software for Windows, p values <0.05 were considered statistically significant.
RESULTS
Cell Proliferation Assays of Fresh and Frozen Chondrocytes from Superficial and Deep Human Knee Articular Cartilage Biopsies
Comparisons were made between fresh and frozen cells from superficial and deep cartilage biopsies, without considering time post-stimulation. In stimulated fresh cells from both superficial and deep cartilage, there was an increase in the proliferative capacity of chondrocytes compared to the non-stimulated controls. After stimulation, a significantly higher proliferative capacity (0.046) was found for fresh chondrocytes from the deep zone biopsies than for those from the superficial zone (0.028) (p<0.05, Mann-Whitney U test) (Fig. 2). The stimulated frozen cells from both biopsy locations also showed an increase in proliferative capacity compared to the non-stimulated controls. Stimulated frozen cells showed no significant difference in proliferative capacity between chondrocytes of deep (0.043) and superficial (0.051) zone origin. There was a significant difference between the proliferative capacity of superficial zone fresh (0.028) and frozen (0.051) chondrocytes (p<0.05, Mann-Whitney U test) (Fig. 2).
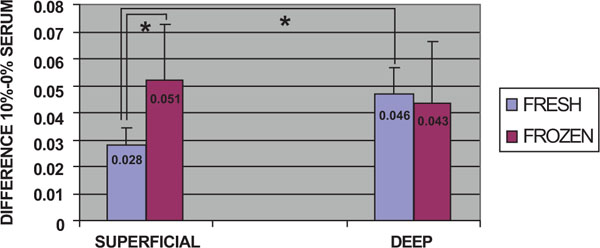
Cell proliferation assays: Comparison of the overall proliferative capacity of fresh and frozen cells related to the superficial or deep origin of the articular cartilage biopsy. Fresh or frozen human knee superficial and deep articular cartilage chondrocytes were stimulated by culturing with Dulbecco's Modified Eagle's Medium (DMEM) with 10% fetal bovine serum (FBS). Following staining with toluidine blue, the cells were solubilized and the absorbance was determined. The results represent the differences in absorbance for stimulated cells relative to the control culture in DMEM without FBS at the same time point. * p<0.05.
mRNA Quantification of Proliferative and Chondrogenic Markers in Fresh and Frozen Chondrocytes from Superficial and Deep Human Knee Articular Cartilage Biopsies
In this study, using quantitative analysis of CCND1 mRNA expression levels by qRT-PCR, the cell proliferation of fresh and frozen chondrocytes from superficial and deep cartilage biopsies were examined. The highest expression levels of CCND1 mRNA were obtained in fresh cells from deep cartilage biopsies (4.36 R.E.L.). Fresh (3.63 R.E.L.) and frozen (3.66 R.E.L.) cells from superficial cartilage biopsies showed equal expression, whereas frozen cells from deep cartilage biopsies showed the lowest expression levels for CCND1 (3.41 R.E.L.). Statistical analyses determined there was no significant differences in CCND1 mRNA expression levels between superficial and deep zone biopsies either from fresh or frozen chondrocytes. There was a trend toward a higher expression of the CCND1 gene in fresh cells from deep cartilage biopsies that did not achieve statistical significance (Fig. 3A).
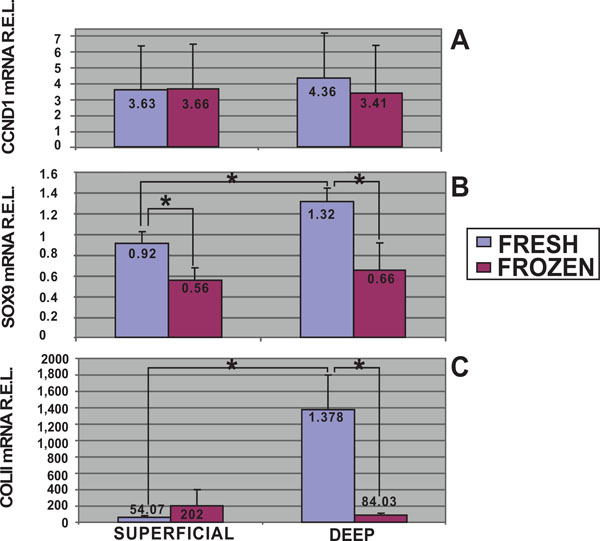
mRNA quantification of proliferative and chondrogenic markers expressed by fresh and frozen cells in superficial and deep origin articular cartilage biopsies. Fresh or frozen human knee superficial and deep articular cartilage chondrocytes were stimulated by culturing with Dulbecco's Modified Eagle's Medium (DMEM) with 10% fetal bovine serum (FBS). qRT-PCR was performed to determine the expression of CCND1 [A], SOX9 [B] and Col II [C] mRNA. The data were normalized against the lowest value obtained for each marker assessed which was assigned the value of 1, and were measured as relative expression levels (R.E.L). *p<0.05.
Moreover mRNA quantitative analyses for chondrogenic markers, SOX9 and Col II, were developed. The highest expression levels of SOX9 mRNA were obtained in fresh cells from deep cartilage biopsies (Fig. 3B). Stimulated fresh cells showed higher SOX9 expression in chondrocytes from deep (1.32 R.E.L.) than from superficial (0.92 R.E.L.) zone (p<0.05, T student test). There was a significant difference between the SOX9 mRNA expression of fresh (0.92 R.E.L.) and frozen (0.56 R.E.L.) chondrocytes from superficial origin (p<0.05, T student test). Regarding chondrocytes from deep cartilage we found statistical significant differences comparing fresh (1.32 R.E.L.) and frozen (0.66 R.E.L.) chondrocytes.
Col II quantitative analyses showed higher expression levels in fresh cells from deep cartilage biopsies (Fig. 3C). In this regard, stimulated fresh cells showed higher Col II expression in chondrocytes from deep (1378 R.E.L.) than from superficial (54.07 R.E.L.) zone (p<0.05, T student test). Regarding chondrocytes from deep cartilage we also found statistical significant differences in Col II mRNA expression comparing fresh (1378 R.E.L.) and frozen (84.03 R.E.L.) chondrocytes.
Analysis of CCND1 Protein Expression Levels of Fresh and Frozen Chondrocytes from Superficial and Deep Human Knee Articular Cartilage Biopsies
To validate the results obtained with cell proliferation assays and qRT-PCR, the protein expression levels of CCND1 in the four different groups of cells were examined (Fig. 4). The highest protein expression levels of CCND1 were obtained from fresh cells from both deep (2.09 R.E.L.) and superficial (1.98 R.E.L.) cartilage biopsies. The values obtained for frozen chondrocytes from deep (1.88 R.E.L.) and superficial (1.76 R.E.L) cartilage were the lowest. These results for CCND1 gene expression for fresh chondrocytes from deep cartilage were confirmed by western blot.
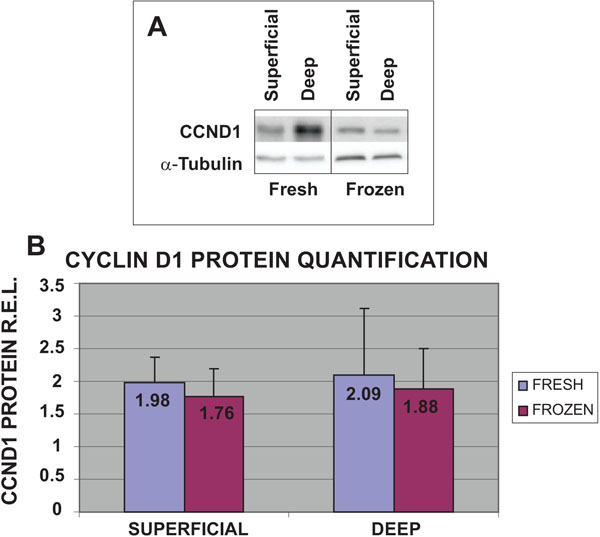
Expression of CCND1 protein in fresh or frozen human knee superficial and deep articular cartilage chondrocytes stimulated by culturing with Dulbecco's Modified Eagle's Medium (DMEM) with 10% fetal bovine serum (FBS). Protein concentrations were measured using a NanoDrop™ spectrophotometer and confirmed by western blot analysis. Data were normalized against the lowest value obtained for CCND1 protein expression levels, assigned the value of 1, and were measured as R.E.L.
Immunofluorescence and Immunohistochemical Analysis of Predictive Markers for Proliferative Capacity of Human Knee Articular Chondrocytes
Histological studies of normal human knee articular cartilage tissue using immunofluorescence and immuno-histochemical analysis showed more positive cells for Ki67 (Fig. 5) in deep (0.59%±0.19) than in superficial (0.11%±0.09) cartilage (p<0.05).
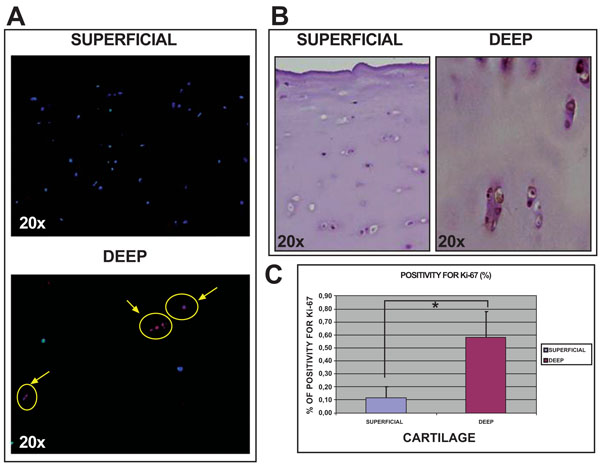
Analysis of Ki67 expression in sections of normal articular cartilage from human knee articular cartilage. Immunofluorescence for Ki67 in both superfical and deep cartilage at 20x [A]. Immunohistochemistry for Ki67 in both superficial and deep cartilage at 20x [B]. Percentage of cells positive for Ki67 in both cartilage biopsies [C]. *p<0.05.
DISCUSSION
Throughout life, articular cartilage is exposed to exogenous mechanical stresses. These mechanical forces on joints can exceed the limit tolerated by cartilage structures, causing erosion with the loss of material from solid surfaces. Additional mechanical factors that can exacerbate cartilage erosion or accelerate its evolution include defective joint structures and functional wear from professional or sport activities, and obesity [39]. To date, most efforts made to repair an articular cartilage injury have been aimed at overcoming limitations of this tissue for healing through the introduction of new cells with chondrogenic capacity [40], or facilitation of access to the vascular system. However, none of the numerous treatments now available consistently regenerates normal hyaline cartilage. Current treatments generate a fibrocartilaginous tissue that differs structurally and functionally from hyaline articular cartilage.
Cell therapy is a new clinical approach for the repair of damaged tissues. A cell-based therapeutic alternative offering more effective repair of focal articular cartilage defects is autologous chondrocyte implantation (ACI) which was developed in a rabbit experimental model [41]. The first clinical application of this method was performed by the group of Brittberg [16], which also demonstrated the successful repair of articular cartilage in rabbits transplanted with autologous chondrocytes [42]. Currently the ACI is proving to be one of the most safe and effective therapies for treatment of focal defects in articular cartilage [7-16, 43, 44]. Cell therapy is most successful in young people, yielding repair tissue of high quality [13, 16]. ACI offers the advantage of using cells that are stably committed to the appropriate phenotype and naturally resistant to vascular invasion, mineralization and ossification [23]. It is used successfully in patients with a particular profile, young people without damage in other joints [45, 46]. ACI is based on the capability of chondrocytes to proliferate in vitro [46]. Previous in vitro studies demonstrated a marked decrease of chondrocyte proliferation and lowered phenotypic stability to be a function of increasing donor age [28, 46], resulting in a tissue that is less able to maintain homeostasis when stressed, leading to a breakdown and loss of articular cartilage, a hallmark of osteoarthritis [47]. The capacity for cell proliferation of chondrocytes is the most important limitation of ACI [7, 18-27, 48]. This proliferative capacity depends not only on donor age, but also on the conditions used to preserve the chondrocytes until the time of implant [49].
This trial was designed to study whether the proliferative and chondrogenic capacity of chondrocytes is dependent on their origin, i.e., superficial or deep cartilage biopsies, and the conditions of their culture and preservation, fresh or cryopreserved. The results demonstrate differences between the proliferative capacities of chondrocytes from superficial and deep articular cartilage biopsies; fresh chondrocytes from deep biopsies showed greater proliferative capacity than fresh chondrocytes from superficial cartilage. These differences were confirmed by CCND1 mRNA and protein detection, which also found increased proliferative capacity of fresh human articular chondrocytes from deep cartilage biopsies. Immunofluorescence and immunohistochemical studies for Ki67 detection found higher protein expression for this marker in deep cartilage. Similar results were obtained for the PCNA marker showing a tendency toward more positivity in deep cartilage than in superficial samples (data not shown). As both markers Ki67 and PCNA are potential predictive markers for proliferative capacity of human articular chondrocytes [28, 29, 50, 51] these results support the idea that chondrocytes from deep zone have higher proliferative capacity. Regarding the chondrogenic potential, stimulated fresh cells also showed higher SOX9 and Col II expression in chondrocytes from deep than from superficial zone. During the isolation and cultivation of chondrocytes for ACI, certain problems may arise, leading to inadequate implantation of these cells, either because there is contamination or because the isolated cells do not proliferate. For additional security, some of the isolated chondrocytes must be cryopreserved. Previous studies [49] demonstrated that the survival and proliferation of chondrocytes were reduced after cryopreservation, particularly evident when the freezing rate was not carefully controlled (-1ºC/min to –40ºC; -2ºC/min to -60ºC; and -5ºC/min to -150ºC). Our study analyzed the proliferative capacity of cryopreserved chondrocytes, using controlled freezing, from both superficial and deep cartilage biopsies. Stimulated frozen cells showed no significant difference in chondrogenic potential between chondrocytes of deep and superficial zone origin. In cryopreserved cells, in contrast to what was observed for fresh cells, there was an indication of increased proliferative capacity in superficial chondrocytes, compared to chondrocytes derived from deep biopsies, although the difference did not achieve statistical significance. An interesting theory that we draw from this study was that cryopreservation selects the most viable and most resistant, and therefore the highest quality chondrocytes, with the freezing and thawing process killing less viable cells. Our results indicate that cryopreservation equally affects chondrocytes from both superficial and deep cartilage biopsies. Both superficial and deep origin frozen chondrocyte populations seem to be homogeneous, showing no statistically significant differences in their proliferative capacity. In contrast, fresh chondrocytes from superficial and deep cartilage biopsies, not subjected to this selective process, were more heterogenous, suggested by the higher proliferative capability of the deep chondrocyte population.
CONCLUSIONS
The highest rates of cell proliferation and chondrogenic potential of fresh chondrocytes were obtained from deep cartilage biopsies. For frozen chondrocytes, there were no significant differences in proliferative and chondrogenic capacity related to biopsy origin. These results indicate that both cartilage biopsy origin and chondrocyte cryopreservation affects the proliferative and chondrogenic potential of chondrocytes and, thus, their ability to regenerate cartilage. Consideration of chondrocyte origin and processing will enhance the success of cell therapy to repair focal articular cartilage injury.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
ACKNOWLEDGEMENTS
This study was supported by grants: Servizo Galego de Saúde, Xunta de Galicia (PS07/84), Cátedra Bioiberica de la Universidade da Coruña and Instituto de Salud Carlos III CIBER BBN CB06-01-0040; Ministerio Ciencia e Innovacion PLE2009-0144; Fondo Investigacion Sanitaria-PI 08/2028 with participation of funds from FEDER (European Community), Tamara Hermida-Gómez is the beneficiary of a contract from Fondo de Investigación Sanitaria (2008), Spain. Emma Muíños-López is supported by the Rheumatology Spanish Foundation, Spain. Silvia Díaz Prado is the beneficiary of an Isidro Parga Pondal contract from Xunta de Galicia, A Coruna, Spain. We would like to thank P Filgueira and MJ Sánchez for technical assistance.
AUTHORS' CONTRIBUTIONS
All authors were involved in the research reported and drafted and approved the final manuscript.

