All published articles of this journal are available on ScienceDirect.

The Epidemiology, Genetics and Future Management of Syndactyly
Abstract
Syndactyly is a condition well documented in current literature due to it being the most common congenital hand defect, with a large aesthetic and functional significance.
There are currently nine types of phenotypically diverse non-syndromic syndactyly, an increase since the original classification by Temtamy and McKusick(1978). Non-syndromic syndactyly is inherited as an autosomal dominant trait, although the more severe presenting types and sub types appear to have autosomal recessive and in some cases X-linked hereditary.
Gene research has found that these phenotypes appear to not only be one gene specific, although having individual localised loci, but dependant on a wide range of genes and subsequent signalling pathways involved in limb formation. The principal genes so far defined to be involved in congenital syndactyly concern mainly the Zone of Polarizing Activity and Shh pathway.
Research into the individual phenotypes appears to complicate classification as new genes are found both linked, and not linked, to each malformation. Consequently anatomical, phenotypical and genotypical classifications can be used, but are variable in significance, depending on the audience.
Currently, management is surgical, with a technique unchanged for several decades, although future development will hopefully bring alternatives in both earlier diagnosis and gene manipulation for therapy.
INTRODUCTION
Syndactyly is a condition well documented both in textbooks and current literature mainly due to it being the most common congenital hand defect.
Coming from the Greek syn (“συν” meaning together) and dactyly (“δακτυλος” meaning digits) it describes an embryological failure of finger separation. Common in some species, including birds and kangaroos, it has a large aesthetic and functional significance for humans born with the condition.
It has long been believed to have a genetic component, and this has been researched greatly over the last decade. Operative management of this condition has historically been, aside a conservative approach, the only definitive care for patients, but are new options becoming available as our understanding of the condition develops?
This article aims to discuss and summarise the current perspective of syndactyly in terms of epidemiology and genetics, as well as considering any new ideas in management of this condition.
SYNDACTYLY: EPIDEMIOLOGY AND CLASSIFICATION
Syndactyly is the fusion of adjacent digits. It is the most common of all congenital hand deformities with an incidence of around 1 in 2000 live births and is twice as common in males, as well as in the Caucasian population [1-4].
Syndactyly may involve fusion of the soft tissues with or without bony fusion. It mainly occurs due to the failure of differentiation between adjacent digits caused by the absence of apoptosis in the interdigital mesenchyme during the seventh and eighth weeks of gestation [1, 5]. The third, fourth, second and first web spaces are affected in decreasing frequency with around 57% of cases occurring in the third web space [2, 3, 6]. The condition is bilateral in half of cases [2, 6].
It is mostly seen in a sporadic appearance, but there is a family history in 10-40% of cases [2, 7]. Inheritance is thought to be autosomal dominant with variable penetrance and expressivity, and this is thought to possibly explain the male predominance [1, 2].
Syndactyly can be an isolated finding or seen with other anomalies such as acrosyndactyly, clindodactyly, synostosis, cleft hand and polydactyly or as a feature in several syndromes including Apert, Poland’s, Pfeiffer, Jackson-Weiss and Holt-Oram.
Syndactyly can be classified in several ways. Anatomically the syndactyly is either simple or complex, and complete or incomplete. Simple syndactyly involves only soft tissues whereas complex includes side-to-side bony fusion. When the adjacent digits are fused all to the finger tip it is described as complete syndactyly, whilst in incomplete refers to only partial union.
The most severe presentation is complex-complicated syndactyly where there is skeletal deformity accompanied by tendon and neurovascular abnormalities; the incidence of which rises as the complexity of the syndactyly increases [2].
Since its first description in the literature, syndactyly has also been classified by its phenotype. The simple and complex, and complete and incomplete are simple anatomical classifications which help to communicating the malformation. The phenotypical classification is more specific for the digits involved, and has led to syndromic and non syndromic syndactylies being described.
Temtamy and McKusick [8] concluded in 1978, from information gathered from both the literature and their own experience, that there were at least 5 phenotypically different types of syndactyly involving the hands, with or without foot involvement. The majority of these are thought to be inherited as autosomal dominant traits. Within each pedigree there is uniformity of the type of syndactyly, allowing for the variation characteristic seen in dominant traits. These genetic forms of syndactyly are required to be analysed separate to syndactyly with congenital amniotic bands for which currently, there is little or no evidence of a genetic basis. This paper will focus on syndactylies involving the hand, and will not discuss lower limb only malformations, or the amniotic band abnormality.
These non syndromic syndactylies appear to only involve digit and appendage malformation, and have since been expanded to nine phenotypes, named syndactyly I to IX, although some are known by their synonyms [9, 10]. See Table 1 for an overview of the nine non-syndromic syndactyly phenotypes.
The Nine Non-Syndromic Syndactyly Phenotypes
| Syndactyly | Sub-Groups | Gene | Loci | Phenotype |
|---|---|---|---|---|
| SD1/Zygodactyly | - | 2q34-q36 | Syndactyly of the 3rd + 4th finger web space and/or the web between the 2nd and 3rd toes | |
| Zygodactyly 1 | - | 3p21.31 | Foot zygodactyly without hand or bony involvement | |
| Zygodactyly 2 | - | - | Bilateral cutaneous and/or bony hand and foot involvement | |
| Zygodactyly 3 | - | - | Specific bilateral webbing, cutaneous or bony, of the third + fourth finger | |
| Zygodactyly 4 | - | - | Bilateral cutaneous webbing of the fourth + fifth toe | |
| SD2/Synpolydactyly | SPD 1 | Homeobox D 13 | 2q31.1 | Syndactyly of the third + fourth fingers associated with polydactyly of all components or of part of the fourth finger in the web. Foot polydactyly of the fifth toe included in a web of syndactyly of the fourth + fifth toes |
| SPD 2 | Fibulin 1 | 22q13.31 | Syndactyly of the third/fourth finger web space and synostosis of the metacarpal and metatarsal bones | |
| SPD3 | 14q11.2-q12 | Third and fourth finger syndactyly with varying degrees of polydactyly of the fourth finger web space. There is also polydactyly of the fifth toe commonly | ||
| SD3 (ODDD spectrum) | Gap Junction Protein Alpha 1 | 6q21-q23.2 | Complete/bilateral, generally soft tissue syndactyly between the fourth and fifth fingers.. The fifth finger is short with absent or rudimentary middle phalanx | |
| SD4/Haas type | LMBR1 | 7q36 | Complete syndactyly, bilateral with polydactyly, generally 6 metacarpals and 6 digits | |
| SD5 | Homeobox D 13 | 2q31-q32 | Soft tissue syndactyly usually affects the 3rd and 4th fingers and 2nd and 3rd toes with associated metatarsal and metacarpal fusion (4th and 5th or the 3rd and 4th) | |
| SD6/Mitten Hand | - | - | Unilateral syndactyly of digits 2-5 | |
| SD7/Cenani-Lenz | LRP4 | 11p11.2 | Severe shortening of the ulna and radius with fusion, fusion of the metacarpals and 'disorganization' of phalangeal development including syndactyly | |
| SD8 | MF4 | ? Xq26 | Fusion of the fourth and fifth metacarpals | |
| SD9/Mesoaxial Synostotic | - | 17p13.3 | completesyndactyly and synostosis of the third and fourth fingers with severe bone reduction in the proximal phalanges, hypoplasia of the thumbs and halluces, aplasia/hypoplasia of the middle phalanges of the second and fifth fingers, and complete or partial soft tissue syndactyly of the toes | |
The syndromic syndactylies, as the name suggests, are linked to other abnormalities in the body and these appear to occur alongside the digit anomalies during foetal development. The list of syndromic syndactylies is extensive and appears to be constantly growing as syndactyly is discovered with additional malformations. In this paper, we will briefly note the genes currently assigned to be causative in the better known and commonly presenting syndromes.
LIMB DEVELOPMENT: GENES AND PATHWAYS
Before discussing the genetics aspects behind each syndactyly, it is useful to understand how the malformation is believed to develop. This paper does not aim to explore the molecular biology of limb formation in detail, but aims to summarise the current findings in a simple, clear, and systematic approach.
The formation of both the upper and lower limbs in the vertebrate appears to be linked to multiple genes as well as a vast number of encoding proteins.
The limb buds and consequent upper and lower limb are formed between the 4th and 8th weeks of gestation and arise from the main trunk, or body. The limb bud is initially directed along three axes of asymmetry, along which the mesodermal cells grow and later become fixed. These axes involve the proximal-distal axis from shoulder to finger, the dorsal ventral axis, from the back to the palm of the hand and the anterior-posterior axis from thumb to little finger. It is this latter axis which appears most important in digit formation. The final and specific limb architecture resulting in the aesthetic limb, normally involves cell proliferation, cell fate determination, cell differentiation and also apoptosis [11, 12].
Two signal centres have been identified to be in control of the human limb structure and positional identity [13-15]. These are the apical ectodermal ridge (AER) which is key for limb growth, and the zone of polarizing activity (ZPA). This latter centre specifies the position and overall patterning in relation to the anterior-posterior axis, although each is dependant on the other [16]. By day 44 the ZPA begins to regress, with metacarpophalangeal joints and proximal phalanges beginning to form. At day 48 the middle phalanges chondrify, followed by the distal bones by day 51 and digit separation by day 54.
This whole process is under the influence of several encoding proteins as illustrated in Fig. (1). In particular the hedgehog pathways, fibroblast growth factors, bone morphogenetic proteins, cartilage derived morphogenetic protein and WNTs have been most discussed in the literature in relation to limb formation [17].

Diagram representing Shh/Gli pathways invloved in limb formation.
Both polydactyly and syndactyly appear to have a relationship with the Hedgehog family of intercellular signalling proteins. These have a majority of functions in cell fate, and most research has involved the Hedgehog (Hh) and Sonic hedgehog (Shh) pathways, specifically in Drosophilia and mice studies [18]. These have particular relevance as Shh is expressed in the ZPA where it controls anterior-posterior limb patterning [19]. In Mice, Shh appears to be a secreted molecule, related to the Drosophilia Hh, which regulates the balance of Gli3 repressor and activator and through these its target genes.
Indian hedgehog (Ihh) is biologically similar to Shh and appears to play a key role in a pathway which is involved in the rate of chondrocyte differentiation regulation [20] and appears to be repressed by fibroblast growth factor receptor 3[21, 22]. Ihh also appears to have a part to play in bone ossification [23]. Multiple papers have suggested a role for the Ihh pathway specifically, in the later development of syndactyly as well as in other congenital abnormalities [24, 25].
The ZPA positioning, and its involvement with Shh, is mainly determined by the transcription factors dHand, Gli3, Alx4 and several Bmp antagonists (Formin and Gremlin). Changes in any of these pathways have been found to lead to the –dactyly malformations (brachy-, syn-, and poly-) [26-30].
Wingless-type MMTV integration site family, members 6 and 10B (WNT6, WNT10B) have both been described as possible genes which require further research due to their involvement in cell apoptosis, and expression in the developing limb bud of the mouse. There has also been noted a relationship to the region of 2q35, a loci hypothesized as the source of syndactyly type 1[31-33].
The WNT family appear to be expressed at a similar time to the fibroblast growth factors (Fgf, in particular Fgf8) in chicken studies [34]. and their pathway is likely to be involved in the final stages of mesenchymal ossification. The Fgf group have been suggested as causative in some of the syndactyly syndromes, and are referenced later in the paper.
The last stage of digit formation involves cell apoptosis, where cell death along anterior, posterior and finally interdigital necrotic zones leads to the familiar profile of the hand [35-37]. This appears to coincide with restriction of Fgf8 expression and down-regulation of Gremlin in these regions [35, 36, 38-40].
Signalling molecules, including the Bone morphogenetic proteins (Bmp) and their antagonist Noggin (Nog) have recently been shown to influence the number of phalanges, possibly having a role in apoptosis [41-47] and blocking their signalling pathway has been shown to result in syndactyly [48-50].
Final digit identity appears dependant on the interdigital mesenchyme. Dahn and Fallon [51] found removal of this in chickens resulted in loss of digit identity, and it appears this is related to Shh and Gli3 pathways [26, 27]. The metalloproteases are also under scrutiny for their involvement in the formation of normal hand architecture, and appear to have a role independent of the Bmp for interdigital web regression [52].
N-Myc and several zinc-finger transcription factors need further research too, as defects in these molecules appear to induce soft tissue syndactyly in mice [53, 54]. Body patterning also appears to be governed by a family of transcription factors which originate from HOX gene encoding. There are 39 HOX genes in the human, as in most vertebrates, organised into 4 clusters and these play an important role in the development of the axial skeleton, central nervous system as well as the urogenital and gastrointestinal tracts, and our main interest, the limbs. There have been links to limb abnormalities with both deletions of some of these HOX clusters (-A and –D) and mutations affecting one or more HOX genes [55].
The specific HOX genes involved in syndactyly will be discussed later in the paper.
CAUSES OF SYNDACTYLY
It should be noted that sporadic syndactyly with no familial history has been documented and environmental factors in-utero that predispose the foetus to syndactyly, as well as other congenital hand abnormalities, have been evaluated. Man conducted a study which reports a probable association with these conditions and maternal smoking [56] and there are suggestions that syndactyly occurrence is associated with lower nutritional and economic status, including increased meat and egg intake whilst pregnant, although more research is required before suggesting these are causative factors [57].
This paper will specifically look at the genetic attributes to syndactyly and we will now discuss the familial form of this anomaly.
SYNDACTYLY: THE GENETIC SEARCH AND A PROBLEM FOR CLINICAL CLASSIFICATION
It has been well documented that syndactyly appears to have an autosomal dominant transmission with variable expression and penetrance [1-4]. This is best represented with the increased prevalence in male offspring, possibly due to reduced penetrance in females. It has also been seen to skip generations and present in a reduced form showing variable phenotype.
This autosomal dominant transmission can be associated either in a simple fashion involving only the distal limbs or as part of a syndrome, some of which will be listed below.
The non syndromic forms of syndactyly are most easily analysed in their subgroups, rather than as one entity and are shown in Figs. (2-10).
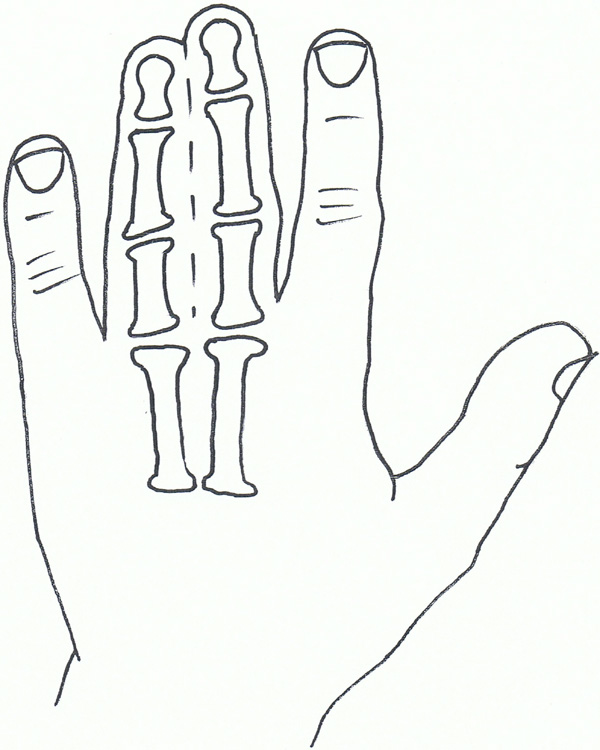
Syndactyly I: Zagodactyly.
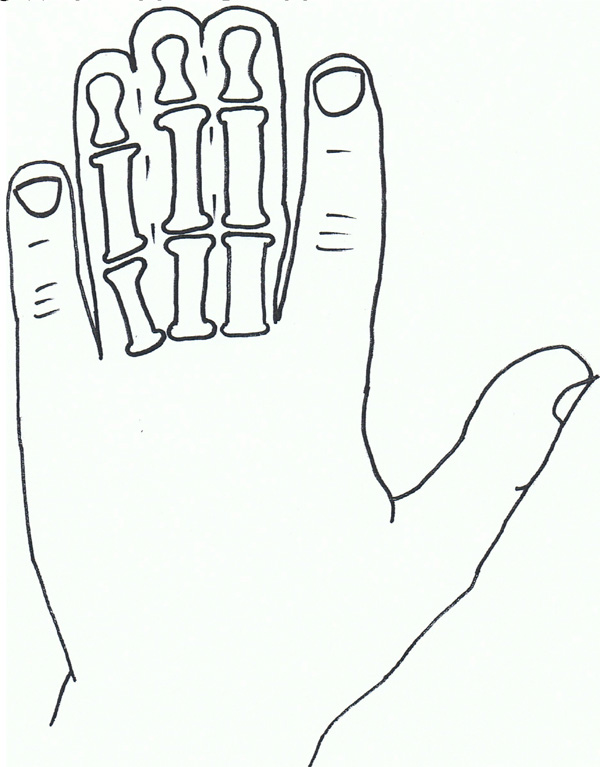
Syndactyly II: Synpolydactyly.
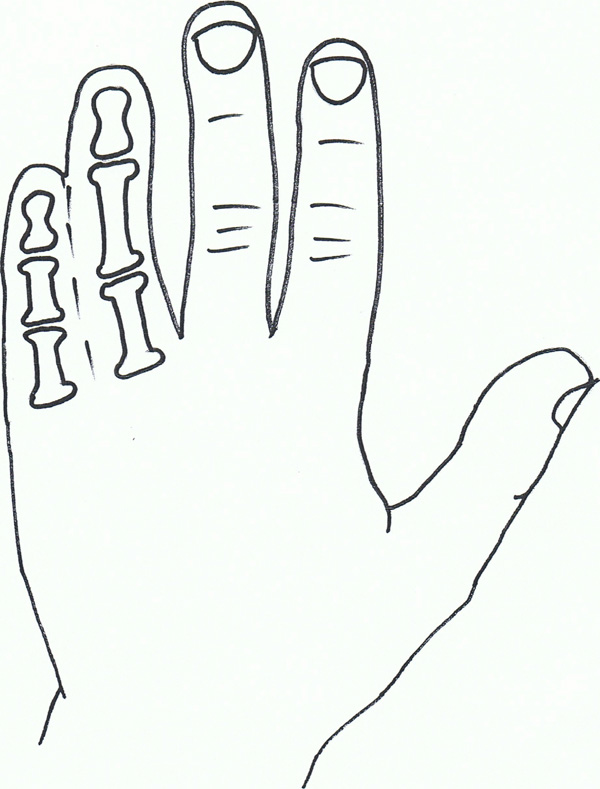
Syndactyly III: Spectrum of oculodentodigital dysplasia.
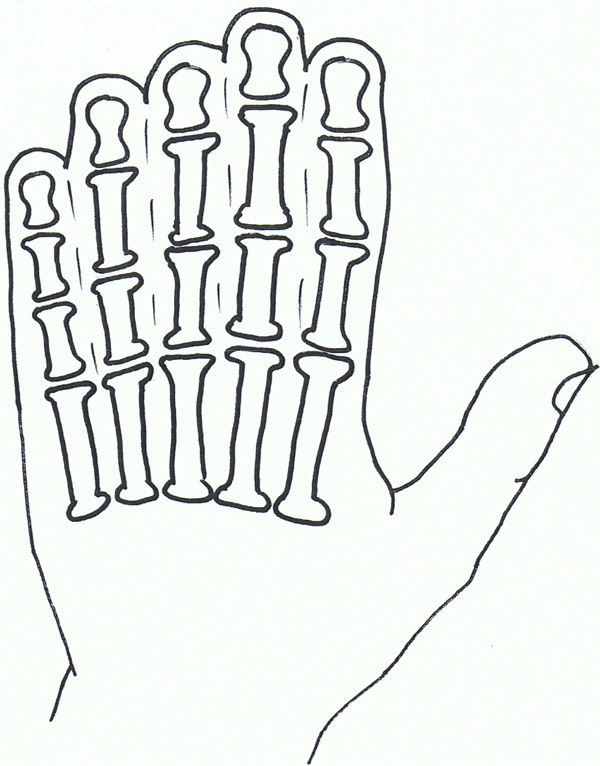
Syndactyly IV: Haas type.
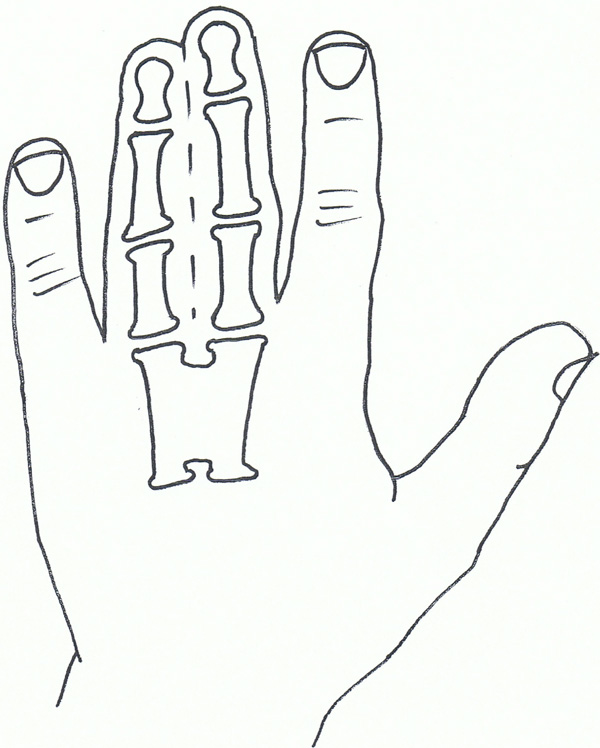
Syndactyly V.
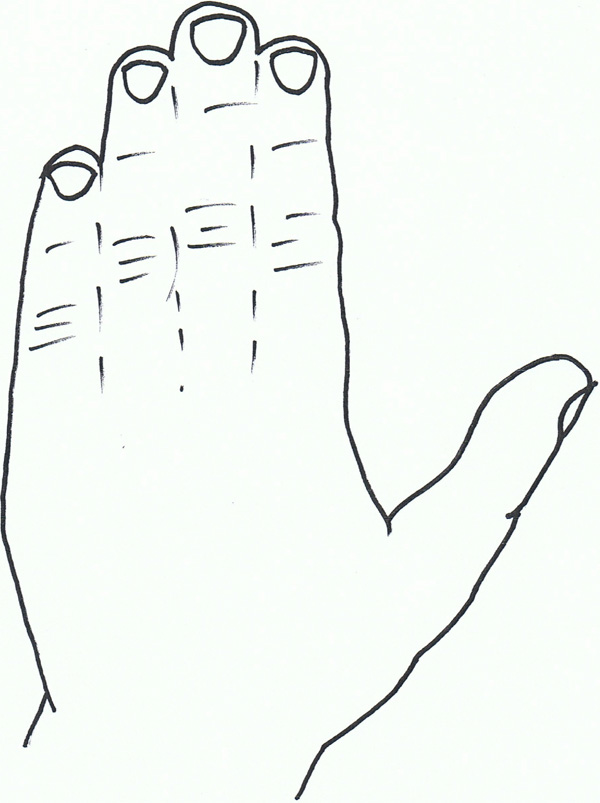
Syndactyly VI: Mitten hand.
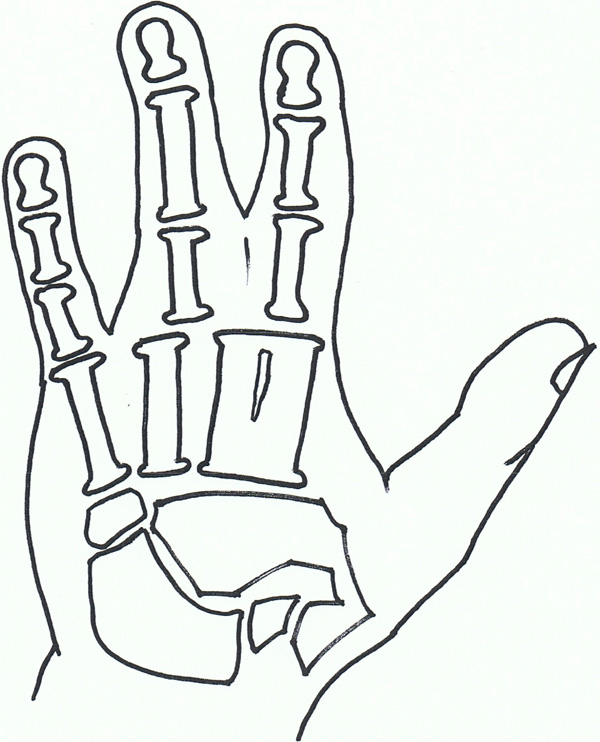
Syndactyly VII: Cenani lenz.
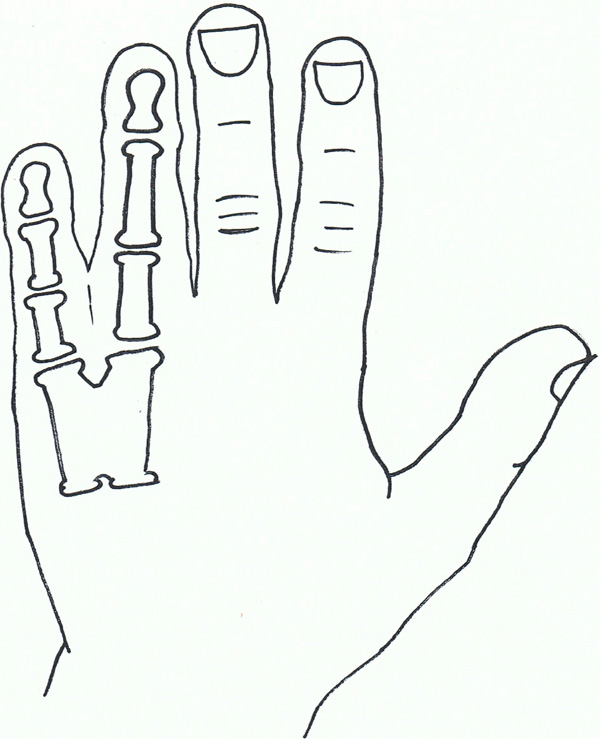
Syndactyly VIII.
Syndactyly IX: Mesoaxial synostotic.
Syndactyly Type I (SD1; MIM 185900)
Also known as zygodactyly, SD1 is characterised by involvement of the 3rd and 4th finger web space and/or the web between the 2nd and 3rd toes. It is the more common non-syndromic presentation of syndactyly and has also been described with involvement of other digits and the underlying bones [58].
The phenotype of SD1 varies greatly. It has been seen to affect the upper or lower limbs, either simultaneously or independently. SD1 appears to be inherited only as an autosomal dominant trait. Initial genetic studies localised the 2q34-q36 region of the second chromosome, mapped during studies involving both a large German and a non-related Iranian family [59, 60]. This loci has also been linked to a Philadelphia type of craniosynostosis with associated syndactyly [61, 62].
Syndactyly Type II (SD2; MIM 185900)
Synpolydactyly (SPD) is, in terms of both genetic and clinical terms, one of the most heterogeneous malformations of the non-syndromic syndactyly types. It also appears to lack penetrance within SPD affected families.
The typical signs of SPD include third and fourth finger syndactyly associated with varying degrees of polydactyly of the fourth finger web space. There is also polydactyly of the fifth toe commonly.
Mouse studies have shown a chemically induced mutation on the mouse chromosome 6 causes syndactyly of digits 2 and 3 of the hind legs (Sndy Jrt/Sndy +). This varies from simple complete to incomplete phenotype, and although sparing the front limbs appears to correlate well with the characteristics of SD1. The homologous region of this chromosomal mutation in humans would be found on 3p25.1 [63].
Malik et al., [64] postulated that SD1 can be further divided into four subtypes;
Subtype 1; Foot zygodactyly without hand or bony involvement,
Subtype 2; Bilateral cutaneous and/or bony hand and foot involvement,
Subtype 3; Specific bilateral webbing, cutaneous or bony, of the third and fourth finger,
Subtype 4; Bilateral cutaneous webbing of the fourth and fifth toe.
They designated the 3p21.31 locus to be specific for this first subtype and named it zygodactyly 1 (ZD1). This appeared to be a new locus for the same phenotype previously described in the German family by Bosse et al.
SPD has been categorised several times in the literature. There is agreement that SPD is inherited in an autosomal dominant manner, and initial subtypes have been differentiated by new findings within the genetic field.
The first genes to be linked to SPD were of the Homeobox family. Particular genes have been located on the 5’ region of the A- and D- clusters of human chromosomes 7 and 2 respectively [41]. These genes appear to influence limb patterning and of particular interest is the Homeobox D gene (HOXD), in particular related to the loci at 2q31 [65].
Research into the HOXD gene mutation has led to the conclusion that in one family this could be further explained by polyalanine expansion in the HOXD13 gene [66-70]. Specifically the N terminal region of the protein, involved in binding to DNA, is disturbed and there appears to be a correlation between expansion size and the appearance and severity of the SPD phenotype in patients, with a greater number of limbs involved with increasing expansion size [71]. It has also been found that minimal duplication does not seem to cause the phenotypical deformity [72]. Since its finding, HOXD13 has been linked with multiple limb deformities including SD type V, brachydactyly and syndromic forms of syndactyly [73, 74].
The initial HOXD13 gene link was supplemented by the discovery of a translocation between Chromosomes 12 and 22 resulting in a defect in the Fibulin gene, which is normally located on the latter [75, 76]. Debeer and Schoenmakers team published further papers examining this translocation within the FBLN1 gene and localised specific involvement of an area represented by EST R72964, as well as ruling out several previously characterised genes [77].
The finding of a further gene, complicated the SPD phenotype further, and so has brought about the widely recognised classification SPD 1-3. SPD 3 correlates to the more classical presentation of SPD and has been linked to the 14q11.2-q12 loci [78].
Likewise, the grouping of gene to phenotype of SPD 2 to the Fibulin 1 gene on Chromosome 12 (MIM 608180) and SPD 1 with Homeobox D13 (MIM 186000) is now widely accepted. SPD2 is generally though to include synostosis of the metacarpal and metatarsal bones.
A more recent paper [79] has stated that SPD should be sub-classed more specifically relating to phenotype, stating genotype- phenotype correlation is weak when looking only at the HOXD13 mutation. They propose the phenotypic variant being classed as (i) typical SPD features, (ii) minor variants, and (iii) unusual phenotypes.
A further subtype aligned to the SPD group is described by one paper [80] where a new distinct clinical form involving a complicated and distinctive hypoplastic synpolydactyly was found. This currently does not appear to have been investigated on a genetic basis, and further research into this will help define this new phenotype as a new or mixed entity.
Continuing research has defined further gene involvement in SPD, although these have been found to involve the Shh pathway on one level or another [81].
Syndactyly Type III (SD3; MIM 186100)
In syndactyly type III, the normal and first described phenotype involves complete and bilateral syndactyly between the fourth and fifth fingers. This is generally a soft tissue syndactyly but occasionally the distal phalanges are found to be fused. The fifth finger is often seen to be short with an absent or rudimentary middle phalanx. The feet are commonly not affected. Johnston and Kirby (1955) [82] presented a family which was one of the largest fully described pedigrees, involving 7 affected males and 7 affected females over 5 generations in a pattern compatible with an autosomal dominant inheritance [8].
Other papers to describe SD3 as a single entity, as opposed to as part of a syndrome include De Smet et al., [83].
Isolated syndactyly type III appears to be in a disease spectrum that includes oculodentodigital dysplasia (ODDD; MIM 164200), which commonly involves digit as well as craniofacial dysmorphia and neurological degeneration [84] ODDD has complete penetrance but a varying phenotype. Gene research has led to the locus 6q21-q23 being associated with SD3, with significant crossover of the locus 6q22-q24, which has been linked to ODDD, and in particular the Connexin 43 (Cx43) gene and its involvement with the Gap junction protein, alpha 1 (GJA1) [85-87].
The Connexin family, consisting of six types, are key in forming gap junctions allowing small molecule and ion passage, and Cx43 is expressed in the developing limb bud with an involvement in causing digit and cartilage condensation [88]. Further studies into both the phenotype and genetic regions above have found localised missense mutations causative for ODDD, of which over 8 have been described, as well as tested in animal studies [89-93].
Specifically, Dobrowolski et al., [94] have shown ODDD phenotype in specific mutations (131M and G138R) whilst mutations at other points result in no syndactyly (H194P) and facial abnormality (G143S). This led to a belief that increased hemi-channel activity may strengthen ODDD phenotype in Cx43 gap junction deficient patients. Other studies have also confirmed a highly variable phenotype of Cx43 mutations which includes ODDD [95-97].
Syndactyly Type IV (SD4; MIM 186200)
Syndactyly type IV is rare, with only four reports in the literature [98-101].
Haas was first to describe this condition and it is commonly known with his name attached (Haas type polysyndactyly). The syndactyly is described as complete, affecting the fingers of both hands, with associated polydactyly, generally involving 6 metacarpals and 6 digits. Flexion of the fingers results in the hands forming a cup-shape. In contradistinction to the type of syndactyly in Apert syndrome [see below], there is no bone fusion. In the reports there was no mention of the condition of the feet, but it is noted there was no associated malformations.
Following an autosomal dominant inheritance trait, 7q36 has been mapped as a locus for SD4 [99]. There is also evidence that mutations of Shh regulation are key in SD4 [102, 103], with one paper showing an involvement of an area of the limb region 1(LMBR1) gene being causative in SD4 development [104].
Syndactyly Type V (SD5; MIM 186300)
This rare form of syndactyly is as a rule characterised by the presence of an associated metacarpal and metatarsal fusion. The metacarpals and metatarsals most commonly fused are the 4th and 5th or the 3rd and 4th. Soft tissue syndactyly usually affects the 3rd and 4th fingers and the 2nd and 3rd toes. Syndactyly is usually more extensive and complete. Kemp and Ravn (1932) [105] described this anomaly in 5 generations of the family from the island of Seeland. There are also descriptions of this syndactyly without metatarsal fusion, but these are usually seen with other foot abnormalities [106].
Syndactyly type V has an autosomal dominant trait but has also been described as X-linked recessive. Research has linked SD5 to the locus at 2q31-q32 as well as mutations in the HOXD13 gene, including the pathogenicity of a c.950A→G (p.Q317R) mutation [73]. This paper also called for a genotype classification of HOXD13 limb morphologies, again confusing the genotype-phenotype boundaries involving the syndactylies.
HOXD13 polyalanine expansion was also found in a study by Kjaer et al., on the described family [107].
Syndactyly Type VI (SD6; MIM Not Allocated)
SD6, also known as mitten hand syndactyly, consists of unilateral syndactyly of digits 2-5 [8]. One family has been described with this anomaly, where an autosomal dominant inheritance, but with variable expression and incomplete penetrance is likely. Tentamy and McKusick included this phenotype in their initial classification, and even today, it is the least researched non-syndromic syndactyly due to its rarity.
Syndactyly Type VII (SD7; MIM 212780)
In 1967, Cenani and Lenz [108] described two brothers with an Apert syndrome-like form of syndactyly. They noted however, that additional features including severe shortening of the ulna and radius with fusion, fusion of the metacarpals and 'disorganization' of phalangeal development were present. The feet of both brothers were less severely affected. They identified similar cases reported by Liebenam (1938) [109], Borsky(1958) [110], and Yelton (1962) [111].
Also named as Cenani-Lenz syndrome, after the pair of doctors, this is a very rare phenotype and has been reported to show an autosomal recessive inheritance. There have been accounts of varying phenotypes, including a description of a patient with features consistent with Cenani-Lenz type but also a severe form of SPD1 [58].
The LRP4 gene has been found to be linked to syndactyly in cattle [112, 113], and is reported, with multiple mutations involving the LRP4 gene on Chromosome 11p12-p11.2, to be the causative factor in SD7 [114]. Of the study group, 2 families did not exhibit LRP4 mutations which suggests further gene involvement. Bachelli et al., had previously found that this gene is unlikely to be related to the pathways involving Formin or Gremlin expression [115], although a more recent paper suggests a mutation involving the loci of these bmp antagonists can result in a phenotype similar to Cenani Lenz syndrome [116].
Within the Cenani-Lenz syndactyly group there appears to be two grossly variant phenotypes- one involving a spoon hand type, the other an oligodactyly type [117].
Syndactyly Type VIII (SD8; MIM Not Allocated)
Fusion of the fourth and fifth metacarpals is also a non-common presentation of syndactyly. First described by Orel in 1928 [118], it was initially thought to have an X-linked recessive trait, which was backed by families described by later papers [119, 120].
Lerch [121] suggested an autosomal dominant inheritance, finding a family with male-male transmission as well as females affected.
Xq26 has been suggested as a starting point for analysis, a known mapped area for split-hand/foot malformation (SHFM2), with the gene allocated as MF4 (MIM309630), although there is general consensus that this syndactyly needs further research [122].
Syndactyly Type IX (SD9; MIM609432)
Type IX, Mesoaxial synostotic syndactyly (MSSD) has been described only in two families. Initially found in a family known to contain SD1, the phenotype of mesoaxial syndactyly, the characteristic features of which were complete syndactyly and synostosis of the third and fourth fingers with severe bone reduction in the proximal phalanges, hypoplasia of the thumbs and halluces, aplasia/hypoplasia of the middle phalanges of the second and fifth fingers, and complete or partial soft tissue syndactyly of the toes, was seen. Percin initially believed this to be a severe form of SD1, with possible homozygous origin [123].
Malik et al., [124] found similar findings in another family, with an autosomal recessive trait, and ruled out genome candidates at 2q34-q36, 2q31, and 6q22-q23. The previous family had had HOXD13 and the genome associated with 2q31 disproved as causative by Percin et al., Merging the two families into one study has revealed a likelihood of a causal gene being mapped to chromosome 17p13.3 [125].
SYNDROMIC SYNDACTYLY
Other presentations of syndactyly are as part of a syndrome, usually with other congenital abnormalities. A brief overview of the key genetic areas in the more common syndromes follows.
Acrosyndactyly, a term for syndactyly associated with congenital constriction bands, appears to lack a genetic basis, with Tentamy and McKusick [8] being first to find little or no evidence of a clear or simple genetic link.
Poland Syndrome (MIM 173800) presents with unilateral hypoplasia or absence of pectoralis muscle with ipsilateral hand and digit anomalies. As of yet no gene or loci have been implemented in its origin.
Acrocephalosyndactyly; there are 5 types of acrocephalosyndactyly, a condition involving syndactyly and craniosynostosis.
Type I, synonymous with the term acrocephalosyndactyly, is Apert syndrome (MIM101200). Associated with the FGFR2 gene, and the loci 10q26, includes mid face hypoplasia, foot and hand syndactyly with a trend for distal bony fusion [126].
Type II, Carpenter syndrome (MIM201000) has been linked to RAB23 gene originating from 6p11, with malformations including foot and hand syndactyly/brachydactyly, and acrocephaly [127].
Saethre-Chotzen syndrome, type III (MIM101400), involves syndactyly of the second and third fingers, as well as the third and fourth toes, as well as eyelid anomalies and cranial abnormalities. It has been linked to the loci 7p21.2 and 10q26 involving the TWIST 1 and FGFR 2 genes respectively [128, 129].
Type IV, was known as Goodman syndrome (MIM2010 20) but is thought now to be a variant of type II [130].
Type V, also known as Pfeiffer syndrome (MIM101600) has been linked to the FGFR 1 and 2 genes [131,132].
Other syndromes and chromosomal location include Acropectorevertebral dysplasia (MIM102510) and 2q36 and Fraser syndrome (MIM 219000) associated with both the sites 4q21 and 13q13, involving the FRAS1 and FREM2 basement membrane genes respectively [133,134], which have also been shown to be linked to fin deformity in zebrafish [135].
Greig cephalopolysyndactyly (MIM 175700) is an autosomal dominant disorder associated with haploin sufficiency of GLI3. This appears to be caused by deletions, truncations or point mutations of the associated Gli3 gene. Similarly the zinc finger domain of Gli3 has been found to be causative in Pallister Hall syndrome whose phenotype includes central nervous system and craniofacial deformities, as well as anal defects [136].
A recent study states a wide range of phenotypes can occur with only a Gli3 mutation, ranging from non-syndromic to syndromic syndactyly [137].
Other genes and loci that have been linked to digital anomalies include the Xq25 loci, with associated developmental delay [138]. Cholesterol metabolism [139], ROR2 [140], nidogen [141], GAS [142] and MBOAT [143] genes have been shown to be related to limb and digit formation in animal and patient groups, likewise mutations in Jagged [144], Serrate [145] and MSX [146] genes appear to cause syndactyly among other congenital abnormalities.
MANAGEMENT OF SYNDACTYLY
The current mainstay for the treatment and management of syndactyly is operative. The indications in general for hand anomalies run true with syndactyly and include;
Functional needs: The degree of function required by the patient; including whether delaying surgery may alter hand function and grip development.
Aesthetic consideration: The appearance of the deformity and its subsequent effects on psychological and social aspects of the patient’s life.
It must be remembered that surgical intervention is not urgent. Despite this there are age related targets for reconstruction and dependency on the type of anomaly present.
Border digit syndactyly involving the thumb, index and ring fingers is felt to benefit from earlier release, usually between the ages of 6 to 12 months, and should always be released first in multiple digit involvement. The result in delayed surgery can involve deformity of the digit relating to forced flexion, angulation and/or rotation.
Long finger syndactyly usually involves similarly lengthened digits and for this reason is often delayed, and KettleKamp and Flatt [147] found results to be better in those aged greater than 18 months, particularly with long term final appearance of the commissure.
Multiple digit involvement, as stated above, should involve in the first instance release of the border digits, with subsequent digit release after a 6-month waiting period. These subsequent releases involve either radial or ulnar sides of a finger as releasing both sides may endanger the digit viability.
It is generally regarded that operative therapy should be done pre-school age. The main reason for this is that interaction with other children whilst having the anomaly is thought to have potentially detrimental affects on the child’s social, functional and psychological development.
The main concern with surgical therapy is the fact that combined circumference of the two normal digits is approximately 1.4 times the circumference of the fused digits. Pre reconstruction it is felt that massaging the skin where the new web space will be constructed facilitates skin loosening and maximises potential soft tissue needed during the procedure.
Full thickness skin grafts are used for soft-tissue coverage in the majority of cases at present, with altering levels of flap use. The choice of flap, and consequent extra need for full thickness graft coverage, appears to be dependant on the surgeon’s preference, with multiple techniques and variances described in the literature. Several other methods have been used, including split thickness grafts [148] and tissue expanders [149]. Both have suffered with less favourable outcomes and increased complications compared to the use of full thickness grafts and have hence failed to establish themselves as alternative techniques. The use of no graft is more common, but this appears only to be of use in mild cases of simple syndactyly.
Follow up should be until skeletal maturity, mainly due to the prevalence of web creep until this age. Complications of all the surgical techniques include web creep, finger deviation (particularly in complex syndactyly) as well as those complications associated with any surgical procedure. The greater the degree of syndactyly, the more need there is for the surgeon to be aware of neurovascular variance and this takes on greater importance in graft and flap survival.
In light of genetic and peri-natal investigations, both these fields have potential for future management input. Ultrasound has allowed earlier diagnosis and this can now be supplemented by genetic review of likely carriers. Constriction bands have been released in-utero allowing a more normal limb development and with minimal scarring [150]. However, in utero surgery at present is high risk and currently the benefit of operating does not outweigh the risks and associated complications for non-fatal conditions.
The continuing development in both the genetic basis of syndactyly, and the improvement in tissue bioengineering, bodes that in the future both these avenues will expand management options.
CONCLUSION
In conclusion, there are currently 9 types of phenotypically diverse syndactyly. This number has increased since the original classification in 1978 by Tentamy and McKusick [8]. The non-syndromic syndactyly are inherited as an autosomal dominant trait, although the more severe presenting types (SD7, SD9 and to a lesser extent SD8) and sub types appear to have autosomal recessive and in some cases X-linked hereditary.
Genetic analysis has found that these phenotypes appear to not be one gene specific, although each phenotype does appear to have its own localised loci, but dependant on a wide range of genes, and subsequent proteins and transcription factors involved in limb formation. The principal genes so far defined in their involvement in congenital syndactyly mainly concern the pathways connected to the Zone of Polarizing Activity and it’s linked Shh pathway.
New syndromes, genes and causative loci are being found as research continues into congenital hand defects and it appears that each new finding gives as many further avenues for further investigation as answers in this field.
Research into the individual phenotypes appears to complicating phenotypical classification as new genes are found both linked, and not linked, to each malformation.
This has been noted by several researchers [151, 152] and attempts have been made to simplify the current classifications although these are yet to be recognised across all specialities.
There is still plenty of research required in the origins and causes behind syndactyly, both as a non-syndromic and syndromic entity, and until this is completed we feel the phenotypical and anatomical description of syndactyly is easiest to learn and use day to day when in clinical practice.
Management is still by surgical correction, and requires optimal planning in terms of patient age and degree of malformation present. This is a non-urgent procedure and it must take into account the psychological and social impact on the child.
Despite increasing knowledge of the causes of syndactyly, management has not changed greatly over the years, although the future is likely to see new techniques involving gene manipulation and tissue engineering at least to be created and studied, although current management is low risk.
CONFLICT OF INTEREST
None declared.
ACKNOWLEDGEMENT
None declared.

