All published articles of this journal are available on ScienceDirect.

Minimally Invasive Treatment of Moderate Lumbar Spinal Stenosis with the Superion® Interspinous Spacer
Abstract
Purpose:
We evaluated the safety and effectiveness of the minimally invasive Superion® Interspinous Spacer (VertiFlex, Inc., San Clemente, CA) in patients with moderate LSS.
Methods:
This single-arm prospective study enrolled 121 patients with moderate LSS between February 2008 and August 2009 and were followed up at 1 (n=111), 3 (n=96), 6 (n=81), and 12 (n=52) months. All patients were treated with the Superion Interspinous Spacer. Main outcomes were back function with the Oswestry Disability Index (ODI), extremity and axial pain severity with an 11-point scale, health-related quality of life with the Physical Component Summary (PCS) and Mental Component Summary (MCS) scores from the SF-36, and adverse events through 12 months.
Results:
ODI improved 64% (p<0.001) through 12 months and clinical success was 92%. Extremity and axial pain improved 53% and 49% (both p<0.001), respectively, through 12 months with clinical success of 76% for axial pain and 86% for extremity pain. Health-related quality of life improved 41% for PCS and 22% for MCS (both p<0.001) through 12 months. PCS clinical success was 81% and MCS clinical success was 62% at 12 months. Four (5.9%) explants were performed although 3 were unrelated to the device. Eight procedure-related adverse events, observed in 6 (5.0%) patients, included superficial incision seroma (n=5), minor wound pain (n=2), and infection (n=1).
Conclusions:
Preliminary results with the Superion Interspinous Spacer suggest that it is an effective and safe treatment option for patients with moderate LSS who are unresponsive to conservative care.
INTRODUCTION
Lumbar spinal stenosis is characterized by narrowing of the lumbar spinal canal and/or the intervertebral foramina resulting from disc degeneration, bulging of the annulus, facet joint hypertrophy, and/or thickening of the ligamentum flavum [1-3], ultimately leading to compression of the neural and vascular elements in the lumbar spine [4]. Neurogenic claudication symptoms such as leg pain and/or weakness during walking result in lower quality of life and impaired functional capacity [5]. With the aging of the population and continuing advances in diagnostic imaging capabilities, lumbar spinal stenosis is becoming more frequently diagnosed with an estimated prevalence of 2 to 13% [6, 7].
Nonsurgical management such as activity modification, pain-relieving and anti-inflammatory medications, physical therapy, and spinal injections are the first-line treatments for patients with mild symptoms although long-term success is marginal, partly because these therapies have no impact on disease progression [8-10]. Consequently, patients are often confronted with the dilemma of living with persistent pain and functional impairment or undergoing invasive surgery, most commonly laminectomy with or without fusion [11-13], a procedure associated with substantial cost and morbidity [14-17]. However, there is no clear treatment algorithm for patients with moderate lumbar spinal stenosis who may obtain only partial relief from conservative measures, but where the severity of symptoms may not justify undergoing invasive surgery such as laminectomy.
Minimally invasive lumbar procedures have become increasingly popular over the past two decades [18] and represent a viable alternative that addresses this therapeutic gap. In particular, interspinous process decompression is a novel procedure that limits back extension at the symptomatic level by implantation of a spacer between contiguous spinous processes. This implant offers the potential to minimize neural injury risk versus alternative procedures and to preserve anatomical structures, thereby affording the option of more invasive surgery in the future should severe symptoms recur. Mid-term results suggest that interspinous spacers improve patient symptoms [19, 20], although long-term safety and effectiveness are currently unknown [21]. The purpose of this study was to evaluate 12-month clinical outcomes in patients with lumbar spinous stenosis who were treated with a new, minimally invasive, interspinous spacer.
MATERIALS AND METHODS
Patients
This single-arm prospective study enrolled 121 patients with lumbar spinal stenosis from Asklepios Krankenhaus (Seligenstadt, Germany) between February 2008 and August 2009. All patients were treated with the Superion® Interspinous Spacer (VertiFlex, Inc., San Clemente, CA). Inclusion criteria for this study included (a) diagnosis of moderate lumbar spinal stenosis, defined as 25% to 50% reduction in lateral/central foramen diameter compared to adjacent levels and radiographic evidence of thecal sac and/or cauda equine compression, nerve root impingement by either osseous or non-osseous elements, and/or hypertrophic facets with canal encroachment, (b) persistent leg, buttock, or groin pain, with or without back pain, that was relieved by lumbar flexion, and (c) unsuccessful conservative treatment for at least 3 months. Exclusion criteria included (a) axial back pain only, (b) grade II to V spondylolisthesis, (c) unremitting back pain in any spinal position, (d) active systemic disease that may affect the welfare of the patient, (e) vertebral osteoporosis or history of vertebral fracture, and (f) pregnant or lactating female. The procedures used in this clinical study were in accordance with the recommendations of the Helsinki Declaration and each patient gave written, informed consent before surgery.
Intervention
The Superion device is a single-piece titanium implant that is delivered percutaneously and deployed between the spinous processes of the symptomatic vertebral levels (Fig. 1a, b). This novel interspinous spacer limits extension at the symptomatic level while preserving mobility, structural elements, and alignment.
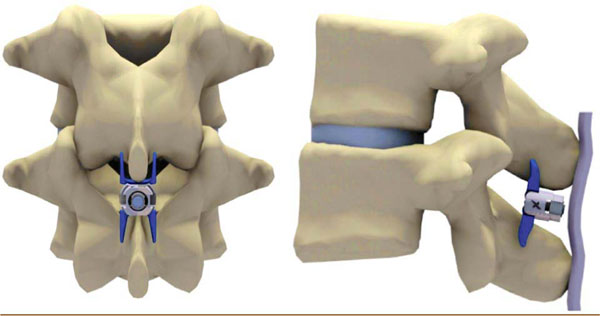
Superion Interspinous Spacer in situ. (a) A/P view, (b) lateral view.
The minimally invasive procedure was undertaken with the patient lying prone on a radiolucent table with the lumbar spine in a neutral or slightly flexed position. Under fluoroscopic guidance or direct visualization, the surgical level was identified and a 12-15 mm midline incision was made. The supraspinous ligament was longitudinally dissected at the symptomatic level and then dilated to ensure adequate room to maneuver within the interspinous space. A cannula was inserted over the dilator and proper alignment and depth were ensured before dilator removal. Next, an interspinous gauge was inserted through the cannula to determine proper implant size selection and final midline positioning was confirmed under fluoroscopy.
The appropriately sized spacer was delivered through the cannula using a device inserter that loaded, inserted into the interspinous space via the cannula, and deployed the implant. Proper device placement was confirmed with fluoroscopy. Finally, the inserter and cannula were removed and the incision was sutured in a standard fashion. Proper placement of the implant is illustrated radiographically in Fig. (2).
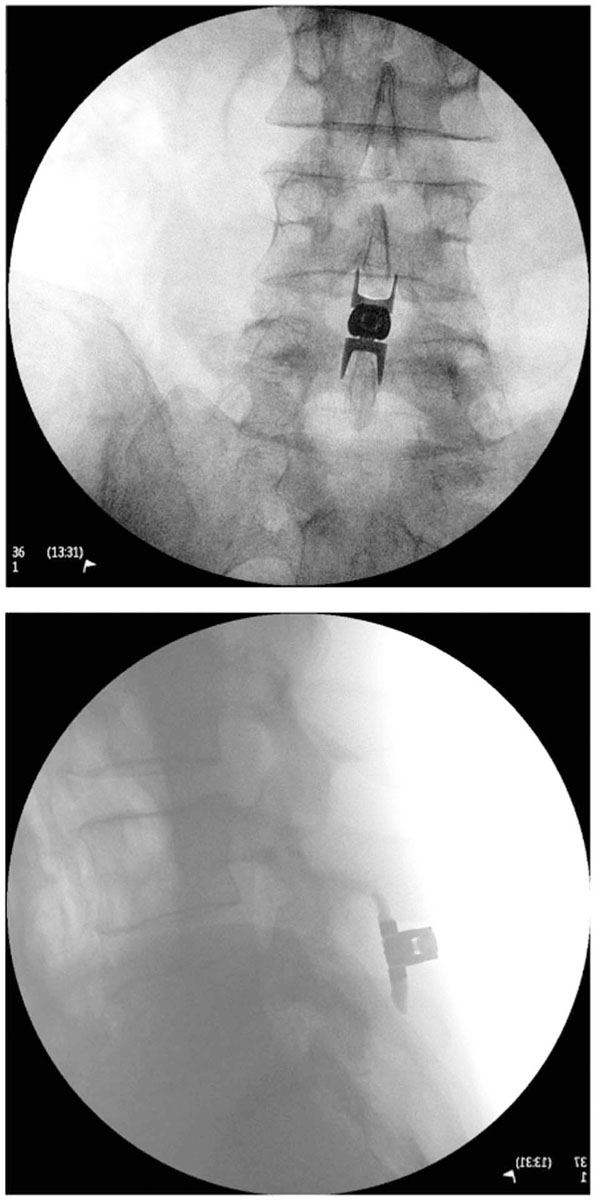
(a) A/P and (b) lateral radiographic image showing a properly placed Superion Interspinous Spacer.
Outcomes
Patients were assessed pre-treatment and then returned for follow-up visits at 1, 3, 6, and 12 months post-treatment. Degree of back-specific functional disability was measured with the Oswestry Disability Index (ODI) (version 2) on a 0 to 100% scale [22]. Extremity and axial pain severity was measured with an 11-point numeric pain scale (0 to 10). Health-related quality of life was assessed with the SF-36 (version 2) and Physical Component Summary (PCS) and Mental Component Summary (MCS) scores were recorded [23]. Safety was assessed by incidence of reported adverse events (AEs) through the 12-month follow-up period. Device-related AEs were defined as implant dislodgement, migration, fracture, or deformation that resulted in clinical sequelae or revision or explant for any reason.
Data Analysis
Data were analyzed using Predictive Analytics Software (v. 18, SPSS, Inc., Chicago, IL). Continuous data were reported as mean ± SD and categorical data were reported as frequencies and percentages. Longitudinal changes in clinical outcomes were assessed with repeated measures analysis of variance. The revision rate over the 12-month follow-up period was estimated with Kaplan-Meier methods. Clinical success was defined as a ≥30% improvement in ODI [24, 25], ≥30% improvement in pain scores [24, 26], ≥5.7-point improvement in PCS [27], and ≥6.3-point improvement in MCS [27], respectively.
RESULTS
The typical patient was aged in the upper 50s, overweight-to-obese, presented with single level disease (most commonly at L4-L5), and suffered moderate-to-severe back and/or extremity pain and functional disability at baseline. Twenty-two (18%) patients presented with concomitant grade I spondylolisthesis. Mean procedure time was 1 hour and hospital stay averaged 3 days. Implant size ranged from 8 to 16 mm with the 12 and 14 mm devices accounting for 80% of implants (Table 1). At the time of this analysis, 52 patients had passed the 12-month follow-up visit window. ODI and pain data were available for 50 of 52 (96%) patients and SF-36 data were available for all patients (52 of 52).
Patient Baseline Characteristics
| Characteristic | Value |
|---|---|
| (n=121) | |
| Age,mean ± SD, y | 57.9 ± 13.5 |
| Female*,n (%) | 60(52.2) |
| Body Mass Index†,mean ± SD, kg/m2 | 29.8 ± 5.0 |
| Treated Level,n (%) | |
| L2 — L3 | 4(3.3) |
| L3 — L4 | 12(9.9) |
| L4 — L5 | 105(86.8) |
| Procedure Duration‡,mean ± SD, h | 1.0 ± 0.2 |
| Length of Hospital Stay§,mean ± SD, d | 3.4 ± 1.1 |
| Axial Pain Severity Score║,mean ± SD | 6.9 ± 1.1 |
| Extremity Pain Severity Score║,mean ± SD | 6.6 ± 1.4 |
| Oswestry Disability Index (ODI),mean ± SD, % | 60.2 ± 7.9 |
* n=115
† n=62
‡ n=110
§ n=102
║ 11-pt.
numeric scale.
Back-Specific Functional Impairment
Rapid improvements in ODI were noted in the first month following treatment (60±8% at pre-treatment to 34±10% at 1 month). Between months 1 and 12, continued improvements in back function were noted with a 12-month mean value of 21±14%, representing a 64% (p<0.001) improvement from pre-treatment levels (Fig. 3). ODI clinical success at 12 months was 92% (46 of 50) (Fig. 4).
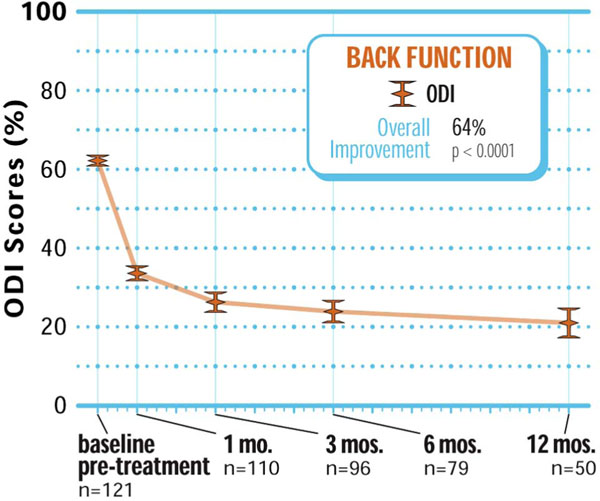
Improvement in back function through 12 months posttreatment.

Back function clinical success rates through 12 months post-treatment.
Extremity and Axial Pain Severity
Extremity pain decreased from 6.6±1.4 at pre-treatment to 3.3±1.4 at 1 month and 2.8±1.5 at 12 months, reflecting a 53% overall improvement (p<0.001) (Fig. 5). Extremity pain clinical success at 12 months post-treatment was 86% (43 of 50) (Fig. 6). Similar improvements were realized in axial pain with values of 6.9±1.1 at pre-treatment, 3.9±1.2 at 1 month, and 3.4±1.5 at 12 months, which represented a 49% improvement compared to pre-treatment values (p<0.001) (Fig. 5). At 12 months post-treatment, 76% (38 of 50) of patients achieved axial pain clinical success (Fig. 6).
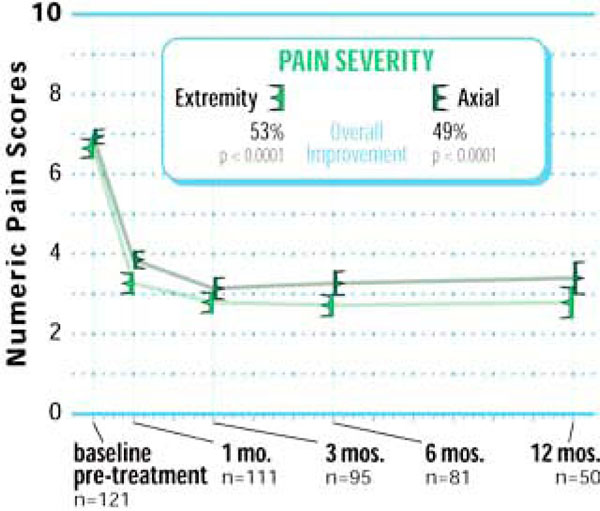
Improvement in pain severity through 12 months posttreatment.
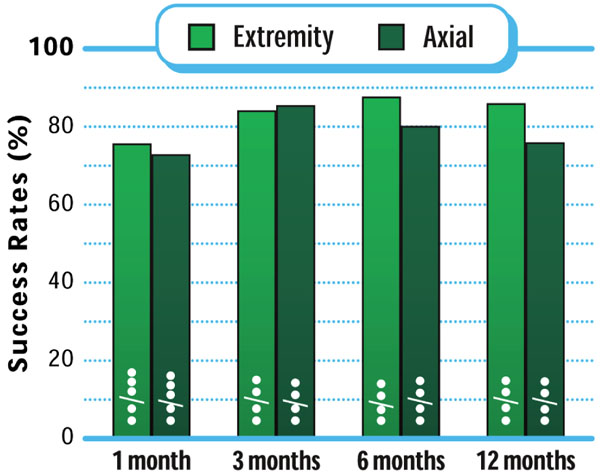
Pain severity clinical success rates through 12 months post-treatment.
Health-related Quality of Life
Six of eight SF-36 domains (PF, RP, RE, SF, BP, VT) improved as early as 1 month post-treatment with all domains showing a significant (all p<0.001) improvement through 12 months post-treatment (Fig. 7). Similar to the trends observed with ODI and axial and extremity pain scores, PCS and MCS each significantly improved from pre-treatment to 1 month with continued improvements observed through the 12-month follow-up visit (p<0.001) (Fig. 8). Through 12 months, MCS clinical success was achieved in 62% (32 of 52) of patients while PCS clinical success, arguably a more clinically important and relevant measure, was achieved in 81% (42 of 52) of patients (Fig. 9).
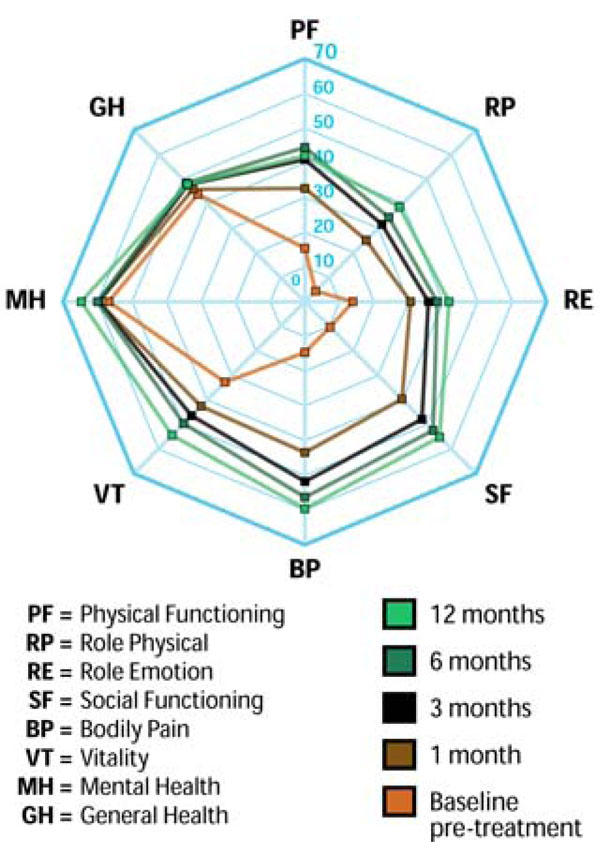
SF-36 assessment of quality of life through 12 months post-treatment.

Physical Component Summary and Mental Component Summary score improvement through 12 months post-treatment.
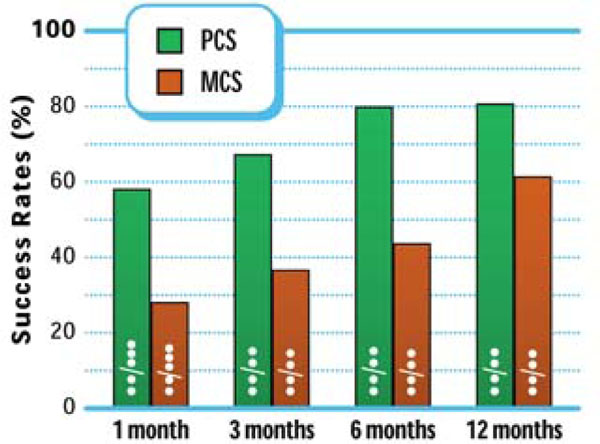
Physical Component Summary and Mental Component Summary success rates through 12 months post-treatment.
Adverse Events
A total of 205 AEs were reported in 121 patients (1.7 per patient) through the 12-month follow-up period. Most AEs were of minor clinical importance and were unrelated to the procedure or the Superion device. Eight procedure-related AEs (5 wound complications, 2 reports of pain, and 1 infection requiring explant on post-treatment day (PTD) 10) were reported in 6 (5.0%) patients. Four patients (5.9%) underwent device explant during the 12-month follow-up period including: (a) an infection-related revision on PTD 10, (b) explant at 2 months due to bulging disk at L3-L4, (c) explant at 8 months due to osteochondrosis, and (d) explant at 11 months due to persistent pain. Postoperative radiographic review noted an incidental case of a slightly angled, albeit correctly placed, Superion device. The patient reported no clinical symptoms and received no treatment associated with this finding.
DISCUSSION
The minimally invasive Superion Interspinous Spacer is a novel, low profile device that results in excellent safety and effectiveness based on the 12-month outcomes reported in the current study including improvements of 64% in ODI, 53% for extremity pain, 49% for axial pain, 41% for PCS, and 22% for MCS. This study represents the first clinical account of patient outcomes with the Superion device.
Patient outcomes following treatment with the Superion device are comparable to those reported with laminectomy, which is the standard of care for surgical LSS treatment. Thomé and colleagues [28] treated 40 patients who presented with symptomatic lumbar spinal stenosis with wide laminectomy. Through 12 to 18 months following laminectomy, pain decreased 45%, PCS improved 38% and MCS improved 24%. However, perioperative complications were reported in 23% of patients and 12% required fusion or adjacent level decompression. These data were corroborated by a meta-analysis that reported a 13% overall complication rate for laminectomy including 0.3% perioperative mortality, 6% dural tear, 3% infection, and 3% deep vein thrombosis [13]. Furthermore, overall success rates with laminectomy are highly variable, ranging from 26% to 100% [3, 13, 29, 30]. Despite similar mid-term patient outcomes, the safety profile of the Superion device suggests fewer potential risks compared to laminectomy.
The Superion device also yields similar, if not slightly better, outcomes as the X-STOP Interspinous Process Decompression System, which is the only FDA-approved interspinous spacer. We reported a 53% improvement in leg pain and a 64% decrease in ODI. For comparison, a study of 175 patients treated with the X-STOP device reported a 36% improvement in leg pain and a 55% ODI improvement through 12 months [19]. Complication rates of 9-12% [31, 32] and a 58% secondary decompression surgery rate within 2 years of implant [33] have been reported with the X-STOP device. The follow-up period in the current study extends only through 12 months so no direct comparison of complication and revision rates can be made with certainty. However, the smaller profile of the Superion device in addition to the less invasive percutaneous delivery of the implant versus the open procedure required for the X-STOP device may yield patient safety advantages (Fig. 10a, b).
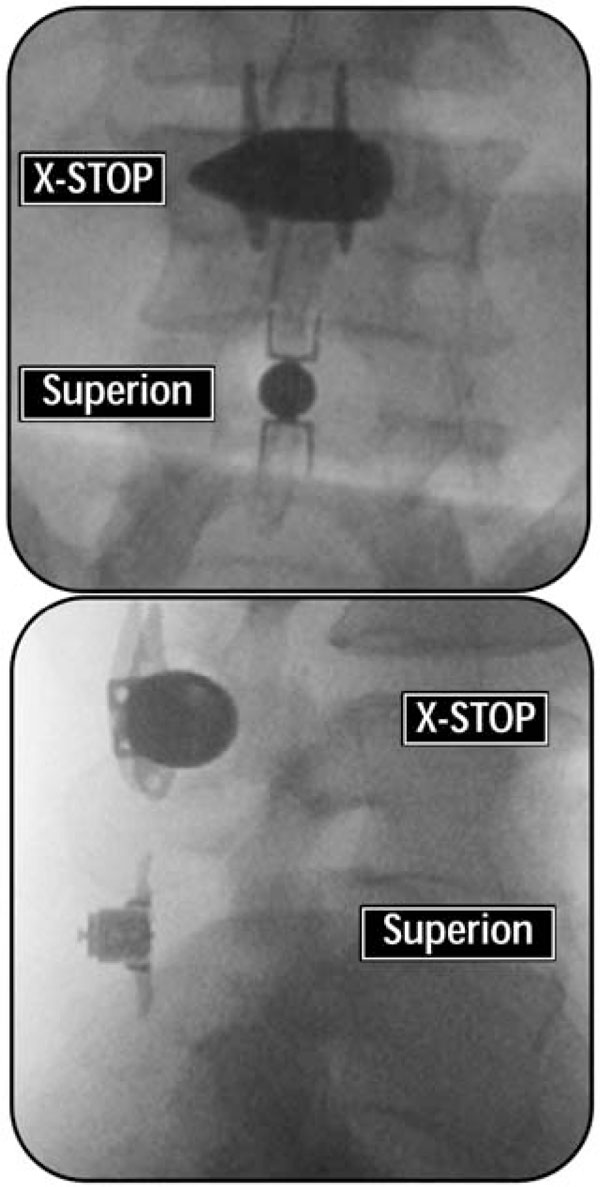
A/P (a) and lateral (b) radiographs comparing the profile of the X-STOP versus the Superion interspinous spacers.
The excellent safety profile of the Superion device for treatment of lumbar spinal stenosis is likely because, unlike laminectomy, implant of this interspinous spacer avoids resection of the posterior spinal elements and thus does not compromise spinal stability and affords a reversible percutaneous device explant if required in the future. A finite element model of a lumbar spine that underwent laminectomy revealed that removal of the posterior elements resulted in increased flexion-extension and axial rotation at the surgical site and the authors concluded that minimization of bone and ligament removal, such as with the Superion procedure, results in greater lumbar stability [34] and potentially lowers risk for fusion surgery. In fact, a cadaver study demonstrated that implantation of the Superion device prevents supraphysiological motion at the symptomatic level and has no adverse impact on the local anatomy [35].
Despite the minimally invasive procedure of interspinous spacer implantation, the average hospital stay in the current study was 3 days. However, this was mainly to support adequate reimbursement under local regulations and did not accurately reflect the convalescence required following the procedure. In fact, an ongoing randomized clinical trial with the Superion device clarifies that implantation should be on an outpatient basis in most cases given the minimally invasive nature of the procedure [36].
The main limitation of this study was the lack of a concurrent control group. Thus, the degree of clinical improvement realized by patients may be biased by non-specific study effects, such as placebo. Regardless, the magnitude of positive clinical outcomes implies a strong beneficial treatment effect of the Superion device. Also, variability in patient follow-up length precludes a definitive assessment of 12-month clinical outcomes. Additional studies of this device are warranted to determine long-term safety and effectiveness outcomes.
CONCLUSION
The Superion Interspinous Spacer is a safe and effective treatment option for carefully selected patients with moderate LSS who are unresponsive to conservative care.
CONFLICT OF INTEREST
Vertiflex, Inc. (San Clemente, CA, USA) provided financial support for development of this manuscript.
ACKNOWLEDGEMENTS
We thank Mr. Randy Asher for assistance with graphical illustrations.

