All published articles of this journal are available on ScienceDirect.

Tissue Engineering Stem Cells – An e-Governance Strategy
Abstract
The rules of governance are changing. They are necessarily becoming more stringent as interventions offered to treat conditions carry unpredictable side effects, often associated with novel therapeutic vectors. The clinical relevance of this relates to the obligations of those involved in research, to ensure the best protection for subjects whilst encouraging the development of the field. Existing evidence supports the concept of e-Governance both in operational health research and more broadly in the strategic domain of policy formation. Building on the impact of the UK Comprehensive Research Network and recent EU Directives, it is now possible to focus on the issues of regulation for cell therapies in musculoskeletal science through the development of the Advanced Therapeutic Medicinal Products (ATMP) category of research products. This article reviews the framework that has borne this and the need for more detailed Virtual Research Integration and Collaboration (VRIC) systems to ensure regulatory compliance. Technology research and development plans must develop in close association between tissue engineering and treating clinicians. The scope of this strategy relates to the handling of human tissues the transport and storage of specimens in accordance with current EU directives and the Human Tissue Authority (HTA) regulations.
‘The things that will destroy us are;
politics without principles
pleasure without conscience
wealth without work
knowledge without wisdom
business without morality
science without humanity
and worship without sacrifice’
—- MK Gandhi [1].
Governance has been aligned with humanity since the dawn of conscience, but with the information revolution has crystallised this into a new form. Governance is defined as ‘the persons who make up a body for the purpose of administering something’ [2].
e-Governance, short for electronic governance [3], uses new information and government technologies to make governance ‘more efficient and more effective’ [4]. Advancement of Information and Communications Technology (ICT) and the implementation of these information services within the public sector increases and improves the operational service provision. This embraces all stakeholders; the government, its citizens, and businesses [5]. E-Governance may thus improve service provision by improving communication between the government and its stakeholders, leading to a more inclusive and holistic modus operandum [6].
In economic theory, such government intervention, seen as correcting ‘market failure’ of a public good or service, is generally the nature of electronic public service provision, and breaks down communication barriers between stakeholders and generates economies of scale [7]. Fig. (1) illustrates the interactions between stakeholders in e-Governance.
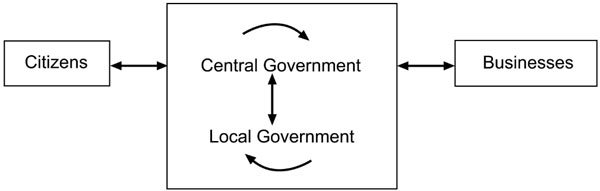
Interactions between groups in e-governance. Adapted after M Bucker [8].
So how prevalent now is e-Governance in the UK? The provision of twenty basic public services online (such as tax, public libraries and car registration) across the European Union (EU) and the total online public service provision averaged 58% in 2007 (compared to 50% in 2006). The UK has approximately 90% of the designated public services available online, whilst Austria leads with 100% provision [9]. So with such an established approach, what role cans e-Governance plays in health research?
The National Institute for Health Research Clinical Research Network (NIHR CRN) aims to improve healthcare in the UK by increasing the efficiency and speed of the translational research process, through providing an electronic infrastructure for clinical research. This infrastructure supports ‘Patient and Public Involvement’ in healthcare (PPI) to encourage more recruits to join clinical trials, and to improve the coordination of the NHS and sponsors (non-governmental organisations) and the private sector [10]. Fig. (2) shows how the broader role of the NIHR as outlined by the Department of Health in their “Best Research for Best Health” publication.
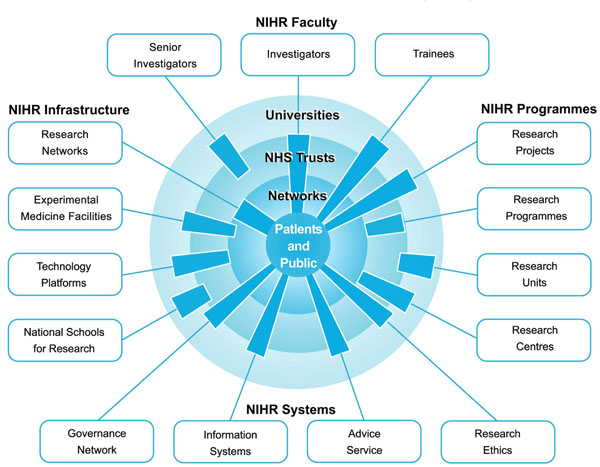
The vision of the NIHR. Adapted from DOH: Best Research for Best Health [11].
NIHR CRN was established in response to the EU Clinical Trials Directive (CTD 2001/20) which was incorporated into British legislation in the Medicines for Human Use (Clinical Trials) Regulations 2004 [12]. This legislation imposed more bureaucracy in clinical trials with the new regulations and conversely it was thought to slow down translational research [13]. In pharmaceutical clinical trials, 70% of trials in phase 2 of testing end because of toxicity or lack of efficacy, and only 20% of drugs entering clinical trials will ever reach the marketplace [14]. Thus it is important that if a trial is to be carried out with only one in five of drugs tested actually becoming licensed, the subjects being tested should be done so under the best possible condition. However there have been some reports that the implementation of the clinical trials directive has slowed and even halted the initiation of clinical research [15]. The application of e-Governance was thus considered a vehicle to overcome these hurdles in the creation of the NIHR comprehensive research network (CRN).
Musculoskeletal medicine is exploiting this infrastructure through an initiative with the NIHR CRN in a model of e-Governance. Facilitated by the Arthritis Research Campaign (ARC) targeted research initiatives in seven topic areas with non-governmental organisations, researchers and industry authorities, led to a programme of clinical trials which will provide a well-defined route for translational research within musculoskeletal medicine [16].
THE RESEARCH GOVERNANCE LEGAL FRAMEWORK
The comprehensive clinical research network (CCRN), launched in 2007 as part of the government’s research and development strategy, ‘best research for best health’, is one of 8 clinical research networks (CRNs) in England forming the infrastructure of the National Institute for Health Research (NIHR). This virtual national research framework is aimed at managing and maintaining the research, research staff and infrastructure of the NHS.
The first ‘CRN’ to materialise was the national cancer research network (NCRN), in 2001. Since then, seven other CRNs developed, guided by the UK clinical research network coordinating centre (UKCRN CC). These CRNs eliminate geographical barriers within the NHS, allowing patients and healthcare professionals from all parts of the country to participate in and benefit from clinical research [17]. The comprehensive clinical research networks (CCRN) were developed to extend the current CRN infrastructure to cover all healthcare topics, facilitating access to NHS patients and research facilities for research within CRNs [18]. It provides a framework, allowing allocation and management of funding for NHS service support, including clinicians’ time, and streamlines the research management process to reduce the burden on researchers. The CCRN is subdivided into 25 comprehensive local research networks (CLRNs), with ‘resident’ populations of about 2 million.
The CCRN forms part of the NIHR portfolio, an information database that ensures all CRN studies are registered, enabling open national competition and peer review. This allows the CCRN, via appropriate CLRNs to provide research management and support services [19].
Information about clinical research can therefore be centralised, making it easier to regulate standards and ensure Good Clinical Practice (GCP). This was defined in the 2001 EU directive as ‘a set of internationally recognised ethical and scientific quality requirements which must be observed for designing, constructing, recording and reporting clinical trials that require the participation of human subjects’ [20]. In the UK, a section of the Medicines and Healthcare products Regulation Agency (MHRA), the GCP inspectorate, is responsible for making sure that clinical trials comply with GCP. The GCP directive requires an approval to be obtained from an Independent Ethics Committee (IEC) before a trial starts. Study design and protocol, investigators, staff and facility quality, consent process, risk assessment, indemnity and payments all require appraisal. Similar to the NIHR portfolio, the EudraCT database was previously set up as part of the GCP directive to share information between member states regarding trial submission dates, ethical approvals and GCP inspections. Whereas the NIHR portfolio regulates clinical trial standards in the UK, EudraCT monitors GCP compliance across all of Europe. This facilitates implementation of directives as information regarding current clinical trials and protocols through one central database.
Greater use of e-Governance has been adopted by several European Union Governments, including the UK [21, 22]. Electronic resources, such as the internet can be used to improve both the administration and consumer (citizen) access to government schemes, resources and services. E-Governance therefore allows consumers to interact with various departments online. Services are delivered in a manner that best suits them and with government citizen interaction streamlining, information can be forwarded to several departments, increasing the efficiency of their interaction [3].
E-Governance for health research administration therefore allows for the support and coordination of otherwise diffuse research projects [23], it simplifies procedures, for initiating and maintaining a research project, such as funding application and patient recruitment, removing many of the barriers to carrying out research of a high quality. This streamlining of processes should ideally place no research project at a disadvantage, regardless of location when accessing resources. Administered by the Comprehensive Clinical Research Network (CCRN) and its allied specialist research networks [23], the operational delivery of the Government’s “Best Research for Best Health” [24] strategy links trial coordinators to the various providers of resources with this framework.
MUSCULOSKELETAL OPERATIONAL IMPLEMENTATION STRATEGIES
It has been argued that the directive, by enforcing stricter regulation has erected a barrier to certain types of research and in some cases may prohibit entirely some trials that could have been carried out pre-directive [25]. The increased bureaucratic burden accompanying the directive has slowed both the workflow of the sponsor and the passage of the trial through the IEC stage. This increased time burden during the preparatory stages stretches the already limited resources of the sponsor. In conjunction with increased insurance costs consequent upon the directive, this has meant that the number of trials initiated has fallen.
This may have both a direct and indirect detrimental effect on patient health since trial drugs often improve the quality of life of participants. Drugs released as a result of such trials are ultimately added to the arsenal of anti-cancer therapies available for the treatment of future patients. Many studies that previously would have been carried out independent of academic grants now require additional funding, by ‘Big Pharma’ resources. It is arguable that the UKCRN and the EU directives work in almost a counter balancing manner. The end result should be though that research can proceed, whilst patient protection and reliable data procurement are ensured.
THE RELEVANCE TO SURGICAL INTERVENTIONS
Trials of surgical procedures would not normally be considered trials of medicinal products and so are not governed by the EU directive but are required to adhere to the Department of Health (DH) Research Governance Framework for Health and Social Care [26]. This is to “ensure that the public can have confidence in, and benefit from, quality research in health and social care”. It is the responsibility of those carrying out the study to ensure that the guidelines are followed – in order that good clinical practice may be achieved. This emphasizes “The dignity, rights, safety and well-being of participants must be the primary consideration in any research study”.
Trials that have separate analysis of treatment effects in children are important, due to their differing physiologies [27] and an increase in trials in children may result to reflect this. It is likely that adult Stem Cell work will need to be satisfactorily completed first.
The Council of Europe defined e-Governance as “a set of technology-mediated processes that are changing both the delivery of public services and the broader interactions between citizens and government” [28]. As the “delivery” of e-Governance has begun to modernise and streamline diverse processes such as researcher-institution interactions. Patient information availability for both clinicians and patients [29] tools has developed to share trial information between clinicians, such as the Orthopaedic Research Base Service (ORBS) and the Virtual Research Integration and Collaboration Network (VRIC) (Fig. 3).

VRIC Trials user interface.
The “Best Research for Best Health” strategy [30], specifically strengthens research processes and collaborations between NHS, the university sector and stakeholders so to facilitate the research of non-NHS clinicians, who have a “direct bearing on the quality of care” of patients under the NHS across different NHS Trusts. The “Research Passport” allows investigators to collaborate across jurisdictions to manage human resources including clinicians’ time [31, 32].
Further streamlining of the process employs an online application system to get NHS permission for clinical trials that fall within the NIHR Clinical Research Network Portfolio within the UK. The IRAS (Integrated Research Application System) provides a single integrated entry point for approval for research for a variety of governing bodies [29];
- Administration of Radioactive Substances Advisory Committee (ARSAC)
- Gene Therapy Advisory Committee (GTAC)
- Medicines and Healthcare products Regulatory Agency (MHRA)
- Ministry of Justice
- NHS / HSC R&D offices (applicable to all trials)
- NRES/ NHS / HSC Research Ethics Committees (applicable to all trials)
- Patient Information Advisory Group (PIAG)
This single access point decreases the time necessary for the Investigator and governing body alike. The European Union Clinical Trials Directive (EUCTD) makes the process “clear and transparent” [33, 34] and dictates how the progress of the research should be monitored, and how practice should be assessed since 2001 [35].
With the National Institute for Health Research (NIHR) Comprehensive Clinical Research Network (CCRN) [36]. Intending to provide a world-wide information base about clinical trials pertaining to any disease and hence display clinical need within the NHS [36], the role of each individual involved in the study is now better defined, so that each may know his exact function in terms of the trial, and so people can be trained specifically.
If there was no waiver of informed consent, then it was not possible to recruit patients into clinical trials in emergency circumstances, such as in intensive care units, but the directive endorses surrogate consent by a “legal representative”; defined by that country’s specific laws to a family member [37]. In spite of these attempts to improve the process, one study also showed that although the number of studies fell by 63% the trial costs actually increased by 85% with insurance doubling from 70 million to 140 million Euros [38].
REGULATION OF CELL THERAPIES – A WHOLE NEW CLASSIFICATION
Many issues must be taken into account. Stem cells themselves remain part of an ethical debate. Adult stems cells (Mesenchymal Stem Cells – MSCs) are widely allowed in research as they equate to, a bone marrow transfusion [39]. Stem cells from already aborted foetuses are more controversial but not as controversial as human embryonic stem cells (hES) taken from early embryos in the blastocyst stage of development [39].
The UK first formulated a law that acted to govern the use of human embryos for stem cell research and the European Science Foundation now collates the various stances of the governments of different countries on this topic [40]. In most cases the source of hES cells has been spare embryos from ‘In Vitro Fertilisation (IVF) treatment and Article 18 of the European Convention on Human Rights and Biomedicine prohibits the creation of all embryos specifically for research purposes [40].
The EU Tissues and Cells Directive for regulation of cell based therapies governs the use of tissues in research and clinical use, including safety and quality of cellular “donation, procurement, testing, processing, preservation, storage and distribution” [41]. The Directive considers stem cell handling, with regard to the stem cell processing when the cells are exposed to the environment provided by a laminar air flow cabinet or a clean room facility [41]. These regulations help to govern cell usage in a way that makes the procedure safer, more cost and time efficient, and with a higher quality standard of practice.
A more recent directive from the European Union is the EU Tissue Directive 2004. This set clear guidelines, standardising stem cell protocols throughout Europe [42]. With regards the donated and processed tissue, it must be tested for infectious diseases and it must be characterised. Patients with allogenic tissue must undergo a thorough medical examination. It may be necessary for the subject to undergo a psychiatric interview to assess whether or not they are mentally fit to undergo a procedure which uses embryonically derived cells. This EU Tissue Directive has been criticised for its lack of clear legal guidelines, leading to widespread discrepancies throughout Europe with regards the laws they adhere to, in terms of their stem cell research [43]. When designing a study for stem cell treatment, such as to evaluate their effectiveness in fracture healing as in the case of the PACINO study, all these regulations must be met for the characterised stem cell type (bone marrow derived mesenchymal stem cells – BMD MSCs).
Again, manifest through the principle of subsidiarity; the Human Tissue Act 2004 regulates the use of human tissue being used in research. The key components of this law relating to cell therapy research are that the researcher must be licensed to carry out these activities and that donor consent must be obtained for the research [44]. As of 2004, a somatic cell therapy being used in research was classified as an investigational medicinal product. These cells are part of research involving ‘Advance Therapy Medicinal Products’ (ATMPs) – regenerative cell therapy products. Obtaining the cells and using them in research must therefore be licensed by the Human Tissue Authority [45].
It is both a legal and statutory requirement that cell-based research adhere to these regulations before the planning of a clinical trial. Under the EU Tissue and Cell Directives Quality and Safety (Q&S) regulations 2007 (implemented as the Human Tissue Regulations 2007) and enforced by the Human Tissue Authority (HTA), cell-based treatment studies on patients are additionally required to take place in institutions with HTA licensure in the procurement, testing, processing, import/export, and distribution of human tissue. The institution is required to meet the standards according to ‘Good Manufacturing Practice’ and three directions detailed by the HTA: 001/2006, 002/2007 and 004/2007 [46].
According to the Medicine and Healthcare Regulatory Agency (MHRA) and HTA, the use of stem cell treatment in this MRC funded double blinded randomised controlled trial of fracture healing using MSCs falls into the definition as an advanced therapy medicinal products/tissue engineering (ATMP). Non-routine applications of ATMPs on individual patients in hospital in accordance with a prescription are even exempted from ATMP regulations. However, exemption of such regulation must continue to follow the HTA Q&S regulations [47].
EARLY CLINICAL STUDIES AND THEIR REGULATION
Musculoskeletal stem cell work involves adult mesenchymal stem cells (MSCs) that are blood, bone marrow [48] and dermally derived [49], though this distinction may not yet be fully appreciated beyond the biomedical community [50]. Stem cells remain controversial since they are unpredictable with respect to their differentiation and amplification. Pluripotent embryonic stem cells also represent a risk of teratoma formation in contrast to adult stem cells, which lack distinct teratogenic potential but remain multipotent [51].
With the EU directives and subsequent National implementation protecting patient welfare, this may restrict the application of MSCs in clinical trials, as they may pose a risk to the patient, similar to the issues surrounding novel gene therapy [51, 52]. It is therefore vital to demonstrate their safety as well as efficacy, classically called phase I and phase II trials.
The EU Tissues and Cells Directive [53-55] were brought into UK law in 2007. It delegated the Human Tissue Authority (HTA) to regulate stem cell lines grown outside the human body for human application [56]. There must be compliance with the established regulations for handling of injectable biological agents. The same is true of the operational procedures derived from this strategic scientific approach.
The process of modification of the initial substrate, either cells or a biologically active agent must be a clearly defined by the ‘end to end’ regulated governing process, to ensure the appropriate compliance with MHRA regulations in the UK, European Medicines Agency (EMA) regulations across Europe and the Food and Drugs Administration (FDA) regulations in the USA. This requires Good Laboratory Practice (GLP), Good Manufacturing Practice (GMP), Good Clinical Practice (GCP) compliance etc, at the various stages of this Translational Research (TR) pipeline.
Quality Assurance (QA) depends on the Standard Operating Procedures (SOPs), often protecting Intellectual Property (IP), since the organisations have invested in developing and refining these processes. They detail the agents and timings for filtering and expanding of cell colonies, maintaining differentiation into specific cell types or de-differentiation to maintain ‘stemness’ with evidence of induction of MSC differentiation for potential clinical applications [57].
Any biological agent should be monitored, including long-term surveillance. This has been promoted as a philosophy within the cartilage community represented by the International Cartilage Repair Society (ICRS) [58, 59] and for a registered UKCRN clinical trial, if adverse events do occur downstream, the association with this trials can be identified [60]. The development of a cell based registry is still awaited. As with arthroplasty, this intervention registry should allow for non-repudiable anonymised data to reconcile with cancer registries in order to either detect early association or ultimately allay fears of inappropriate cell transformation which underlies any type of biological intervention.
CONCLUSIONS
What might first appear to be a regulatory barrier is simply a series of highly complex filters providing checks and balances to ensure risk mitigation for trial subjects. Certain ‘entry level’ criteria are therefore essential before considering embarking on even on collaboration with such work. Such organisation’s governance processes should consider who the stakeholders are in this process and what specimens need to be monitored. When records need to be kept and reviewed and where specimens can be held and the standards of facilities required. It is vital to understand why such processes are to be monitored and reported and how this will be delivered locally.
Anyone involved in either the clinical collection of specimens or their handling, both in transit and in the laboratory in the organisation, needs to be aware of this and those directly involved should be GCP certified.
All specimens which are collected from human subjects either for research in a non-GLP facility or processed in the GLP tissue fabrication facility need to be identifiable, though anonymised before transfer for basic science work unless it is specifically part of the protocol, for example in trials where GMP facilities are to be used and cells replanted.
Records need to be held by each institution, and reconciled with each other in order to ensure reliable and consistent tracking of tissue specimens. It is not essential that the records are completed immediately, due to the more pressing matters such as ensuring preservation of the specimens, but certainly the records should be updated within a working week of any tissue transfers. Modern bar-coding can ease most of these administrative tracking duties.
These are then available for review by the licence holders, or their designated individuals. Such records are a legal requirement for the organisations, and as such, should be available for inspection by authorities such as the medicines and Healthcare products regulatory agency (MHRA) and the Human Tissue Authority (HTA). These organisations notify in advance in writing of any inspections, but best practice should ensure that records are kept up-to-date.
Part of this process is the development of the concept of corporate governance. This involves integration of components of research, clinical and information governance. These policies need to be integrated, in the same way as with research pipelines; main areas of good laboratory practice (GLP), good clinical practice (GCP) and good manufacturing practice (GMP) interrelate. Other areas of regulation that does not impact directly here, are good pharmacovigilence practice (GPvP) and good delivery practice (GDP) but are dealt with by the pharmacy.
By linking adequate facilities (fit for purpose) directly, so that specimens are handled and logged in and out these facilities, traffic between operating theatres and the Institutional laboratories is monitored. Once specimens are handled within the laboratory, it depends whether specimens need to be handled in accordance with good laboratory practice (GLP) processes or whether the experiments on cells do not require this. It is therefore necessary if the specimens are to be handled in accordance with GCP and GLP requirements that there are tissue transfer agreements in place for any such trials.
In order to facilitate integration, it is sensible to employ a single system for the tracking of tissue specimens that can link to the trials registry as well as in the virtual research integration and collaboration environment (VRIC). This should cover the interface with the clinical services and thus the jurisdiction of the clinical HTA licence holder. Inclusion of the GLP and non-GLP facilities remains under the jurisdiction of the research HTA licence holder. Such processes are likely to be reformed in future, as recommended by the Academy of Medical Sciences report [61] which proposes ‘A new pathway for the regulation and governance of health research’. The only certainty is change, and administrations need to be able to accommodate this. E-Governance is likely to be the most effective method, but needs a different approach that is better metered to the perceived risks and is imposed by government per se, rather self governing.
The adoption of a database is only part of the solution for the operational delivery of this strategic e-Governance policy. Staffs need training to accurately record specimens in the system and also monitor the transport across sites. This should be as straightforward as possible, but fully compliant with all regulation to be reviewed at least biannually.
It is only when the entire process of ‘top-down’ and ‘bottom-up’ management of such trials is reconciled that society will be able to reflect on whether these reforms have achieved their original goal of improving safety and efficacy of such trials for these advanced therapeutic products and society can comfortably move forward with the clear conscience that regulation is in place but the necessary burden does not stifle innovation.
CONFLICT OF INTEREST
None declared.
ACKNOWLEDGEMENT
None declared.

