All published articles of this journal are available on ScienceDirect.

Effect of Cytoskeletal Disruption on Mechanotransduction of Hydrostatic Pressure by C3H10T1/2 Murine Fibroblasts
Abstract
Cyclic hydrostatic pressure of physiological magnitude (< 10 MPa) stimulates chondrogenic differentiation of mesenchymal stem cells, but mechanotransduction mechanisms are not well understood. It was hypothesized that an intact cytoskeleton would be required for uninhibited mechanotransduction of hydrostatic pressure. Therefore we examined the effects of drugs which selectively interfere with actin and tubulin polymerization on pressure-induced upregulation of aggrecan and col2a1 (type II collagen) mRNA expression. C3H10T1/2 cells were cultured as pellets in either 4µM cytochalasin D or 4µM nocodazole and subjected to 3 days of cyclic hydrostatic compression (1 Hz, 5 MPa, 2 h per day). Phalloidin staining and indirect immunostaining with anti α-tubulin antibody confirmed disruption of microfilament and microtubule assemblies, respectively. Real time RT-PCR revealed that both drugs substantially lowered the basal level of aggrecan and col2a1 mRNA, but that neither drug prevented a pressure-stimulated increase in gene expression relative to the altered basal state. Thus upregulation of macromolecular gene expression by cyclic hydrostatic pressure did not require a completely intact cytoskeleton.
INTRODUCTION
It has been shown that intermittent application of 0.1-10 MPa hydrostatic pressure stimulates chondrogenic differentiation of mesenchymal stem cells, as evidenced by increased mRNA expression of genes such as aggrecan, type II collagen, and Sox9 and increased production of cartilaginous extracellular matrix macromolecules [1-6]. However, the mechanism(s) of hydrostatic pressure mechanotransduction is not well understood. Typically occupying 16 - 21% of a cell’s volume [7] and linked to extracellular matrix elements, the cytoskeleton is often implicated in mechanotransduction of cellular distortion (e.g. stretch). However, its role in transduction of hydrostatic pressure has not been fully characterized. Because hydrostatic pressure can be a major factor in chondroinduction of mesenchymal stem cells, it was proposed to investigate the importance of the cytoskeleton in transduction of hydrostatic pressure by C3H10T1/2 murine fibroblasts, a model of primary bone marrow mesenchymal stem cells. Any cell undergoing division will have an altered cytoskeleton, and the cytoskeleton itself may become a target for therapeutic agents designed to interfere with cell division (e.g., anti-cancer drugs). Thus this area of research may have basic science and clinical relevance.
In primary bovine chondrocytes subjected to 5 MPa of cyclic hydrostatic pressure the actin stress fibers resembled those of non-pressurized controls. However, as the amplitude of cyclic loading was increased to 15 and 30 MPa, the structure of actin fibers was severely affected by the pressure while no change was detected in staining patterns of the intermediate filament vinculin [8]. It has also been reported that nocodazole and taxol, a microtubule depolymerizer and stabilizer, respectively, both prevented hydrostatic pressure-induced stimulation of proteoglycan synthesis in chondrocytes [9]. Higher magnitudes of continuous hydrostatic pressure (≥ 24 MPa) substantially suppressed proteoglycan synthesis [10] and caused major changes in the distribution of actin and tubulin similar to those typical of osteoarthritic chondrocytes [11].
It was hypothesized that an intact cytoskeleton would be essential for hydrostatic pressure to elicit an upregulation of cartilage marker gene mRNA by C3H10T1/2 murine fibroblasts. We adopted a conventional approach of using drugs that depolymerize specific cytoskeletal components. Cytochalasin D, a drug that inhibits actin-filament polymerization and can affect cell shape and deformation under stress [12, 13], was used to investigate the importance of the actin microfilaments. Cytochalasin D collapses the actin filament network and destroys microfilament organization. However, it does not significantly alter tubulin (microtubule) or vimentin (intermediate filament) architecture. The importance of microtubules was investigated using nocodazole, an antineoplastic agent which selectively depolymerizes microtubules by binding to tubulin. The purpose of this study was to determine the effects of cytochalasin D and nocodazole on hydrostatic pressure-induced upregulation of aggrecan and col2a1 mRNA by C3H10T1/2 murine fibroblasts. The C3H10T1/2 cell line is capable of undergoing chondrogenic, osteogenic, myogenic, and adipogenic differentiation [14-17], and the differentiation pathway is strongly influenced by the culture conditions. Chondrogenesis is favored when cells are grown at high density in the presence of transforming growth factor beta-1 [18] or bone morphogenetic protein-2 [19].
METHODS
Cell Culture
C3H10T1/2 mouse embryonic fibroblasts clone 8 cells (CCL-226, American Type Culture Collection, Manassas, VA) were propagated in standard 75 cm2 polystyrene tissue culture flasks. Unless indicated otherwise, culture medium and supplements were from Sigma (St. Louis, MO, USA). Growth medium contained high glucose (4,500 mg/l) Dulbecco’s Modified Eagle Medium (DMEM), 10% fetal bovine serum, and 1% antibiotic/antimycotic solution. Prior to reaching confluence, cells were subcultured using trypsin. In accordance with the donor’s recommendation that the line be used between the 5th and 15th passages, cells between passage 5 and 7 were trypsinized and resuspended at 5×105 cells/ml. One milliliter aliquots of this suspension were added to 1.5 ml-microcentrifuge tubes and pellets formed by brief (~ 10 s) centrifugation at 10,400 rpm. After allowing 48 h for pellet consolidation at 37ºC, pellets were transferred to 2 ml screw-top glass vials containing growth medium with 25 ng/ml recombinant human bone morphogenetic protein-2 (Wyeth Pharmaceuticals, Cambridge, MA, USA) and 50 μg/ml ascorbic acid and incubated for an additional 24 h. Drugs were then added (see Experimental Design) and the vials sealed with flexible, PTFE-lined silicone rubber septa (P. J. Cobert Associates, St. Louis, MO, USA), so as to exclude all air. Each vial contained 2 or 3 pellets.
Experimental Design
Vials were assigned to one of the following treatment groups: Control (no load, no drug), CSK Disrupted (no load, with drug), Pressure (with hydrostatic pressure, no drug), and Combined (with hydrostatic pressure, with drug). Vials in the CSK Disrupted and Combined groups contained either 4 μM cytochalasin D or 4 μM nocodazole, whereas vials in the other 2 groups contained the same medium without drug. Vials in the Pressure and Combined groups were subjected to cyclic hydrostatic pressure for 2 h on each of 3 consecutive days: a 1 Hz sinusoidal waveform with consistent minimum and maximum magnitudes of 0.4 and 5 MPa. The experiment was repeated once and the data pooled such that 6 independent samples (2 pellets per sample) were analyzed for mRNA expression. The custom pressure chamber consisted of a domed, flanged stainless steel base and lid bolted together to compress a Teflon® gasket [20, 21]. The water-filled chamber was pressurized via a flexible hose connected to a hydraulic cylinder mounted in a servohydraulic testing machine (Bionix 858, MTS, Minneapolis, MN, USA). During loading vials in the Control and Drug groups were placed in a separate non-pressurized water-filled chamber. Both chambers were immersed in the same 37ºC water tank. Temperature was regulated via CSC32 digital controller (Omega Engineering Inc., Stamford, CT, USA) with thermocouple feedback and 500W immersion heater. Water in the tank was continuously circulated by a 230 gallon/h submersible pump.
Total RNA Isolation
Immediately after the endpoint of the entire 3-day loading session, all vials were removed from their respective chambers and two cell pellets from each vial were placed immediately in 1 ml of RNA stabilization solution (RNAlater™, Qiagen Inc., Valencia CA, USA). For those glass vials containing three pellets, the third pellet was reserved for cytoskeletal imaging. Total RNA was isolated using the RNeasy® Mini kit (Qiagen Inc., Valencia, California, CA, USA). Concentration and purity of the eluted RNA was measured by reading absorbance at 260 and 280 nm in a spectrophotometer (SPECTRONIC 601, Bausch & Lomb, Rochester NY or ND-1000 Spectrophotometer, NanoDrop Technologies, Wilmington DE).
Primer Sequence and qRT-PCR
PCR primers (Table 1) for mouse glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a housekeeping gene and those for aggrecan and collagen, type II, alpha 1 (col2a1) were designed using Primer3 (v 0.4.0) online software [22]. Real Time quantitative reverse transcription polymerase chain reaction (qRT-PCR) was carried out to quantify the relative mRNA expression of genes of interest in experimental groups relative to controls. Thermal cycling and real-time detection of fluorescent cDNA was performed in an iCycler iQ® (Bio-Rad, Hercules, CA, USA). Reverse transcription and reverse polymerase chain reactions were performed using the iScript One-Step RT-PCR kit with SYBR® Green (Bio-Rad, Hercules, CA, USA). Thermal cycling parameters were as follows: 10 min at 50ºC for cDNA synthesis, 5 min at 95ºC for iScript reverse transriptase inactivation, 10 sec at 95ºC and 30 sec at 55ºC for PCR cycling and detection for 40 cycles. Then, melt curve analysis composed of 1 min at 95ºC, 1 min at 55ºC and 10 sec at 55ºC, was performed to confirm specificity of product. Load- and drug-induced fold changes in the genes of interest were calculated using the ΔΔCT method [23], and data were statistically analyzed by two-way ANOVA at the 95% confidence level (α = 0.05).
Sequences of Primers Used for qRT-PCR
| Primer | Sequence | Product Size | Genbank Accession Number |
|---|---|---|---|
| GAPDH | (forward) 5’-CTGAGGACCAGGTTGTCTCC-3’ (reverse) 5’-GCCTCTCTTGCTCAGTGTCC-3’ |
226 bp | M32599 |
| COL2A1 | (forward) 5’-GCCAAGACCTGAAACTCTGC-3’ (reverse) 5’-GCCATAGCTGAAGTGGAAGC-3’ |
239 bp | NM_031163 |
| Aggrecan | (forward) 5’-CTCAGTGGCTTTCCTTCTGG-3’ (reverse) 5’-CTGCTCCCAGTCTCAACTCC-3’ |
185 bp | L07049 |
Cytoskeletal Imaging
Pellets to be used in cytoskeletal imaging studies were equilibrated in buffer PEM (100 mM PIPES, pH 6.9; 1 mM MgCl2; 1 mM EGTA) with 4% polyethylene glycol (MW 40,000). They were then fixed for 2 h in PEM containing 3% formaldehyde followed by 1 h permeabilization in PEM containing 0.5% Triton X-100 and 30 min digestion in 1 mg/ml hyaluronidase in PBS at 37º. For visualization of microfilaments, pellets were stained for 1 h with Alexafluor 488-labeled phalloidin (Molecular Probes, Eugene, OR, USA) diluted 1:40 in PBS containing 1% bovine serum albumin. For visualization of microtubules, indirect immunostaining with anti α-tubulin antibody was performed. Pellets were incubated overnight at 4ºC with monoclonal anti α-tubulin antibody (clone DM1A, Sigma-Aldrich, St. Louis MO, USA) diluted 1:500 in PBS with 1 % BSA. They were then rinsed extensively in PBS and incubated for 1 h in fluorescein isothiocyanate-conjugated secondary IgG antibody diluted 1:25 in PBS. Both phalloidin and immunostained pellets were thoroughly rinsed in PBS to reduce background and were then examined by confocal laser scanning microscopy.
RESULTS
Pressure generated within the chamber was calculated by dividing the force (as recorded from 25 kN load cell) by the hydraulic cylinder bore area of 958 mm2. The minimum and maximum applied hydrostatic compressive stresses of 0.4 and 5 MPa were maintained consistently over the 2 h duration of cyclic pressurization at 1 Hz. Glass vials in which the cultures were sealed contained only very small, if any, entrapped air bubbles, so that pressure was transmitted instantaneously to the inside of the vials. The MTS actuator stroke necessary to achieve the desired pressure was approximately 15 mm. At the end of a loading session, the temperatures inside the experimental and control chambers were within 0.2º C of the external bath, which was maintained at 37 ± 0.2 ºC.
Gene Expression
All melt curves exhibited a single sharp peak indicating the presence of primer-specific amplicons. The difference between CT values of technical replicates was always less than 0.4. The GAPDH housekeeping gene’s CT value was 12-14, and the standard deviation of biological replicates was less than 0.5. CT values for Aggrecan and col2a1 were in the range of 18-25, and standard deviations of biological replicates were less than 1. Fold changes in mRNA expression are reported as mean±standard deviation (Figs. 1-3). In the absence of cytoskeleton-altering drugs, cyclic hydrostatic pressure caused a 2.26 ± 0.71-fold upregulation of aggrecan mRNA (p = 0.0028) and a 1.72 ± 0.51-fold increase in col2a1 mRNA (p = 0.0055) (Pressure vs. Control). Treatment with either drug alone had the opposite effect. Aggrecan mRNA was downregulated by 0.67 ± 0.19-fold (p = 0.0048) and col2a1 by 0.34 ± 0.11-fold (p = 0.0003) in response to treatment by cytochalasin (CSK Disrupted vs. Control). Nocodazole decreased aggrecan mRNA by 0.35 ± 0.22-fold (p = 0.0007) and col2a1 mRNA by 0.27 ± 0.17-fold (p = 0.0003) (CSK Disrupted vs. Control). However, neither drug significantly diminished the pressure-induced upregulation of either aggrecan or col2a1 above the drug-lowered baseline. In the presence of 4 μM cytochalasin-D, hydrostatic pressure induced a 1.54 ± 0.42-fold increase in aggrecan mRNA and a 2.07 ± 0.56-fold increase in col2a1 (Combined vs. CSK Disrupted). These fold increases were not statistically different from those induced by Pressure relative to Control. Similarly, pressure increased aggrecan mRNA by 1.79 ± 0.75-fold and col2a1 mRNA by 1.64 ± 0.30-fold in the presence of 4 μM nocodazole (Combined vs. CSK Disrupted). These effects were not statistically different from those produced by Pressure relative to Control.
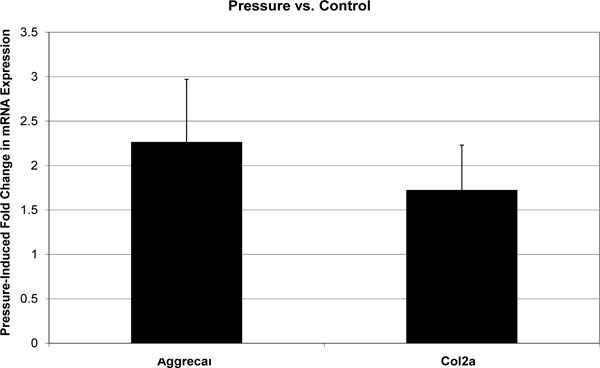
Effect of cyclic hydrostatic pressure on mean aggrecan and col2a1 mRNA expression in C3H10T1/2 murine fibroblasts. Each error bars is one standard deviation. Control is represented by a fold change of 1. Pressure had a statistically significant effect on the expression of both genes relative to Control.
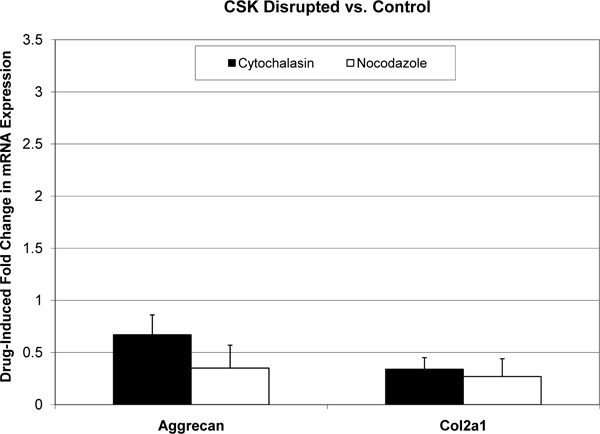
Effect of cytoskeleton-disrupting drugs on mean aggrecan and col2a1 mRNA expression in C3H10T1/2 murine fibroblasts. Each error bars is one standard deviation. Controls are represented by a fold change of 1. Cytochalasin-D and nocodazole had statistically significant effects on the expression of both genes relative to Control.

Effect Effect of cyclicd hydrostatic pressure on mean aggrecan and col2a1 mRNA expression in presence of CSK-disruptive drugs. Each error bars is one standard deviation. The CSK-Disrupted baseline is represented by a fold change of 1. Pressure had a statistically significant effect on the expression of both genes relative to this CSK-Disrupted baseline. These effects were similar to those of Pessure relative to Control (in the absence of CSK-disruptive drugs).
Visualization of MFs and MTs under Laser Scanning Confocal Microscopy
Phalloidin staining and indirect immunostaining with anti α-tubulin antibody were carried out to visualize microfilaments and microtubules, respectively (Figs. 4, 5). Each drug had a profound effect on its cytoskeletal target. Specificity of the drugs towards these targets had been confirmed in a pilot study (i.e. no apparent effect of cytochalasin D on microtubules or of nocodazole on microfilaments). Control cells displayed prominent actin stress fibers and a fine filamentous network of microtubules. These structures were abolished by cytochalasin-D and nocodazole, respectively. Actin and tubulin appeared to be collapsed into globular clusters. Pellets subjected to hydrostatic pressure were more firm and did not flatten against the glass slide as much as the controls; hence the somewhat smaller and more circular but there were no other apparent effects of pressure on microfilaments or microtubules. Likewise, drug-treated pellets which had been pressurized had a similar appearance to their non-pressurized counterparts.
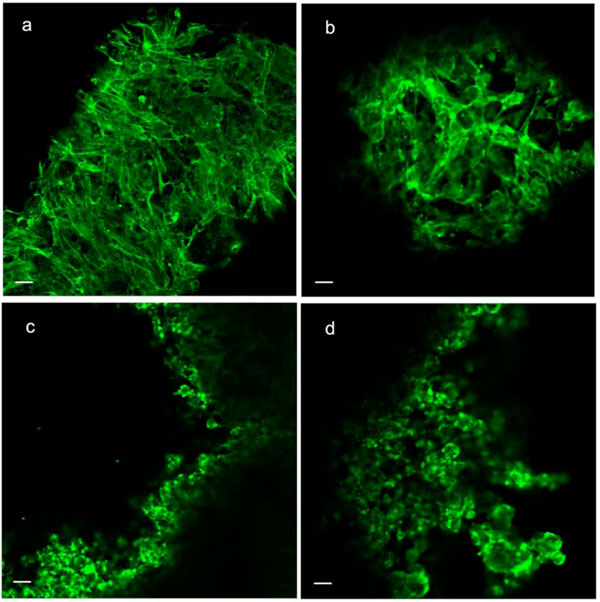
Phalloidin staining of microfilaments in C3H10T1/2 cell pellet. (a) Control; (b) Pressure (5 MPa hydrostatic pressure applied at 1 Hz for 2 h); (c) CSK Disrupting (4 µM cytochalasin D); (d) Combined (pressure + cytochalasin). Scale bars = 25µm..
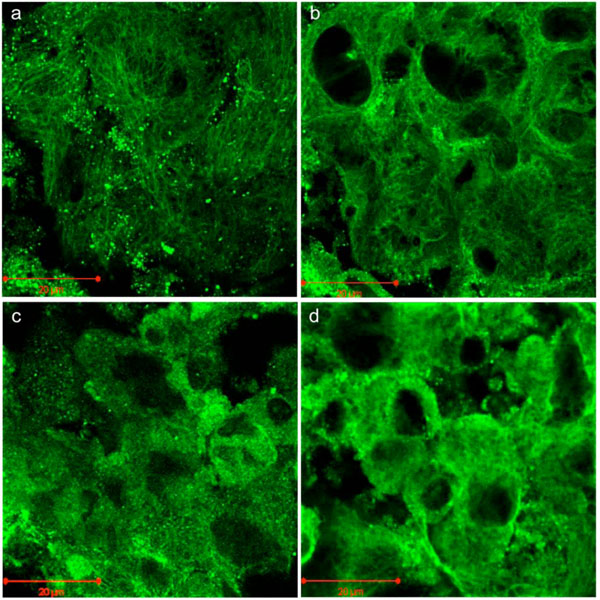
Indirect fluorescent immunostaining of microtubules in C3H10T1/2 cell pellet. (a) Control; (b) Pressure (5 MPa hydrostatic pressure applied at 1 Hz for 2 h); (c) CSK Disrupting (4 µM nocodazole); (d) Combined (pressure + nocodazole). Scale bars = 20µm.
DISCUSSION
Tibiofemoral joint contact pressure is in the range of 3-11 MPa [24], over 90% of which is supported by interstitial fluid pressure [25, 26]. Therefore much attention has been focused on hydrostatic loading as a means of promoting chondroinduction of mesenchymal stem cells. Indeed, many studies have confirmed that cyclic hydrostatic pressure can enhance chondrogenesis by primary bone marrow mesenchymal stem cells and C3H10T1/2 murine fibroblasts [1-6]. Cyclic pressurization to 0.1 MPa is sufficient to enhance chondroinduction [27], and higher pressure magnitudes elicit a stronger response [28]. The prevailing theory of cartilage mechanobiology is based on the concept that intermittent hydrostatic pressure is the greatest stimulus for cartilage maintenance [29], and in vitro experiments have confirmed that intermittent hydrostatic pressure increases chondrocyte marker gene expression, macromolecule production, and extracellular matrix deposition by mesenchymal stem cells [1-6]. However, the cellular mechanisms by which mesenchymal stem cells transduce hydrostatic pressure remain largely unknown. The purpose of our study was to determine the importance of the cytoskeleton in mechanotransduction (transformation of an applied physical stimulus into a cellular biomolecular response) of hydrostatic pressure by C3H10T1/2 murine fibroblasts, a model for primary bone marrow mesenchymal stem cells. Cells undergoing division, such as may occur in a healing osteochondral defect, a cell-laden tissue engineering scaffold, or a bony fracture gap, will have an altered cytoskeleton. In addition, some anti-cancer drugs, such as vinblastine, work by interfering with cytoskeletal function. Knowledge gained through studies such as this may enhance our understanding of cartilage tissue engineering, fracture healing, potential anti-cancer drug side effects, and the etiology of osteoarthritis.
C3H10T1/2 cells were used as a model of bone marrow mesenchymal stem cells because of their homogeneity, rapid growth rate, low biosafety risk, and low cost. Previous studies of mesenchymal stem cell mechanobiology have examined the effects of hydrostatic pressure in the physiological range of 1-10 MPa applied at or near typical gait frequency of 1 Hz [1,3,4,6], and a similar loading regimen was utilized in the current study. Mechanotransduction was based on upregulation of aggrecan and type II collagen mRNA. Aggrecan and type II collagen are the most important structural components of articular cartilage and together they account for approximately two-thirds of its dry weight. Therefore they are widely used as markers of chondroinduction and chondrocyte biosynthetic activity. More importantly, the mechanosensitivity of aggrecan and type II collagen mRNA is well established, with substantial increases observed for both in response to hydrostatic pressure [3,6,21,30,31].
The cytoskeleton is known to mediate force transfer from the extracellular matrix to the nucleus [32] and has been implicated in mechanotransduction of several kinds of distortional stress, including shear [33], stretch [7], and compression [34]. But hydrostatic compression, pressure acting with equal magnitude in all directions, will not produce a change in cell shape; rather, it will tend to decrease the cell’s volume. The cell can be considered to behave as a biphasic viscoelastic solid, with water as one phase and the cytoskeleton and organelles together making up the other. Such a structure is predicted to undergo minimal volumetric strain under hydrostatic loading. Although a decrease in chondrocyte volume has been observed under distortional compressive loading, this decrease was attributed to fluid exudation rather than compressibility of cellular components and cytoplasm [35], as Wilkes and Athanasiou [36] had previously reported on the intrinsic incompressibility of osteoblast-like cells.
Our study examines the effect of disrupting either microfilaments or microtubules on the hydrostatic pressure-induced increase in macromolecular gene expression in C3H10T1/2 murine fibroblasts. We had previously tested the effects of cytochalasin D and nocodazole on the C3H10T1/2 microfilament and microtubule networks, respectively, and determined that 4 μM was sufficient to achieve thorough disruption, as we observed in the present study. The selective action of these drugs has been verified by other investigators [37, 38]. The combination of cytochalasin and nocodazole was somewhat cytotoxic, so experiments were not performed in the presence of both drugs. We found that treatment with either cytochalasin D or nocodazole drastically lowered aggrecan and col2a1 mRNA expression, which highlights the importance of the cytoskeleton in maintaining normal cell function.
Concerning the effects of nocodazole, our results are consistent with previous findings that it inhibits proteoglycan production by articular chondrocytes [9, 10]. But in our study cells exposed to either drug responded to cyclic hydrostatic pressure of physiological magnitude (5 MPa) by increasing expression of both genes above the drug-suppressed baseline. Furthermore, the magnitude of the increase was approximately the same as that observed in drug-free cells relative to drug-free controls. Our data contrast somewhat with those of Jortikka et al. [9]. They showed that microtubules mediated the proteoglycan biosynthetic response to 5 MPa cyclic hydrostatic pressure, as no stimulation of proteglycan secretion occurred in monolayer chondrocytes treated with nocodazole. There are many differences in the two models which could account for the apparent disparity, including the type of cells (transformed mesenchymal stem cells vs. primary articular chondrocytes) and culture configuration (monolayer vs. pellet). In addition, cellular strain due to deformation of the substrate is a possible confounding factor in their experiment. It is also possible that nocodazole does not inhibit a cells’ ability to perceive the hydrostatic pressure and respond from a transcriptional standpoint, but does interferes with its ability to increase synthesis or secretion. In chondrocytes, nocodazole treatment was associated with fragmentation and dispersion of the Golgi apparatus [10], where glycosylation and sulphation of proteoglycans occurs. In a pattern similar to that which we observed, Bianchi et al. [39] found that actin- and microtubule-perturbing toxins inhibited EAAT3 glutamate transporter activity but did not prevent the phorbol ester-stimulated increase in transport from this lower basal activity level.
Future studies will be designed to determine if cytoskeleton perturbation interferes with the pressure-induced activation or mobilization of intercellular signaling molecules upstream of gene transcription. In summary, cytochalasin-D and nocodazole, which selectively disassemble microfilaments and microtubules, suppressed aggrecan and col2a1 mRNA expression in C3H10T1/2 cells. However, neither drug alone prevented the hydrostatic pressure-stimulated increase in macromolecule mRNA expression relative to the lowered basal level.
ACKNOWLEDGEMENTS
This research was supported by the Mississippi State Bagley College of Engineering. Technical support and equipment were provided by the Mississippi State Electron Microscope Center and the Mississippi State Life Sciences and Biotechnology Institute.

