All published articles of this journal are available on ScienceDirect.

Osteocompatibility of Biofilm Inhibitors
Abstract
The demand for infection prevention therapies has led to the discovery of several biofilm inhibitors. These inhibiting signals are released by bacteria, fungi, or marine organisms to signal biofilm dispersal or disruption in Gram-positive, Gram-negative, and fungal microorganisms. The purpose of this study was to test the biocompatibility of five different naturally-produced biofilm chemical dispersal and inhibition signals with osteoblast-like cells: D-amino acids (D-AA), lysostaphin (LS), farnesol, cis-2-decenoic acid (C2DA), and desformyl flustrabromine (dFBr). In this preliminary study, compatibility of these anti-biofilm agents with differentiating osteoblasts was examined over a 21 days period at levels above and below concentrations active against bacterial biofilm. Anti-biofilm compounds listed above were serially diluted in osteogenic media and added to cultures of MC3T3 cells. Cell viability and cytotoxicity, after exposure to each anti-biofilm agent, were measured using a DNA assay. Differentiation characteristics of osteoblasts were determined qualitatively by observing staining of mineral deposits and quantitatively with an alkaline phosphatase assay. D-AA, LS, and C2DA were all biocompatible within the reported biofilm inhibitory concentration ranges and supported osteoblast differentiation. Farnesol and dFBr induced cytotoxic responses within the reported biofilm inhibitory concentration range and low doses of dFBr were found to inhibit osteoblast differentiation. At high concentrations, such as those that may be present after local delivery, many of these biofilm inhibitors can have effects on cellular viability and osteoblast function. Concentrations at which negative effects on osteoblasts occur should serve as upper limits for delivery to orthopaedic trauma sites and guide development of these potential therapeutics for orthopaedics.
INTRODUCTION
Orthopaedic implants may be composed of a variety of different materials, such as metal alloys and polymers, which provide non-natural surfaces in the body and are a haven for biofilm formation [1]. Attachment of bacteria to medical device surfaces, such as orthopaedic implants, leads to the formation of a biofilm, which may cause life-threatening infections and healthcare cost burdens [2]. Orthopaedic infection is the second most common complication with large joint replacements and is very difficult to diagnose and treat [2, 3]. The consequences of infection include loss of implant function, damaged local tissues, and, in extreme cases, septic shock and death [4]. Treatment usually requires multiple surgeries and aggressive courses of systemic and local antibiotic therapies, with accompanying risks and side effects [3, 5].
Bacteria communicate within biofilm communities with chemical signals to trigger various events, such as detachment from a surface into the planktonic form when resources are scarce, a process known as quorum sensing [6, 7]. The planktonic form of bacteria can be 10-1,000 times more susceptible to antibiotics than a colony protected by a biofilm [8]. Some of the different types of bacterial- and fungal-produced signals that disperse biofilms in Gram-positive, Gram-negative, and fungal microorganisms are D-amino acids (D-AA), lysostaphin (LS), farnesol, and cis-2-decenoic acid (C2DA) [9-1]. In addition, Desformyl-flustrabromine (dFBr) is a marine-derived molecule that has been shown to have anti-biofilm activity against multiple bacterial strains [12]. Combining these biofilm dispersal and inhibition agents with antibiotics can greatly increase the susceptibility of bacterial biofilm to antibiotic therapy [13].
While the antimicrobial effects of these biofilm disrupting agents have been characterized, little is known about their effects on osteoblast cell function. The presence of these chemical signals could slow down or halt the healing process depending on the toxic concentration released at the site [14]. Therefore, the purpose of this study is to test the biocompatibility of the aforementioned five different anti-biofilm agents with osteoblasts in vitro at levels above and below active concentrations through assessment of viability and differentiation over a 21 day time course compared to controls.
MATERIALS AND METHODS
Farnesol, D-AA (D-phenylalanine, D-proline, and D-tyrosine), dFBr, and LS were purchased from Sigma. C2DA was purchased from Grupo Nitrile.
MC3T3 mouse calvarial osteoblast cells (ATCC) were seeded at 1 × 104 cells/cm2 in 24 well plates in alpha-MEM containing 10% fetal bovine serum (FBS) with antibiotics, 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B. After overnight attachment, media was replaced with osteogenic media consisting of alpha-MEM with 10% FBS, 0.1 μM dexamethasone, 0.2 mM ascorbic acid 2-phosphate, 10 mM beta-glycerophosphate, 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B. Farnesol, C2DA, and an equal mixture of the three D-AAs were dissolved and diluted serially in 1.25% ethanol to improve solubility of these biofilm inhibitors with hydrophobic characteristics. LS and dFBr were solubilized and diluted in osteogenic media including antibiotics. Solutions of each chemical in alpha-MEM or alpha-MEM + ethanol were added to achieve the concentrations listed in Table 1 and a final ethanol concentration of 1.25% for those with added ethanol. Osteogenic media alone, osteogenic media + ethanol, and 10% FBS in alpha-MEM (non-osteogenic) were also evaluated as positive and negative controls. Media was refreshed every 3 days. At days 1, 3, 7, 14, and 21 cells in wells (n=4 per group per time point) were lysed with 25 mM Tris and 0.5% Triton X-100 and stored at -80°C until analysis. Cell number was estimated by DNA quantity using Quant-it™ PicoGreen (Invitrogen), and alkaline phosphatase (ALP) levels were determined through a colorimetric assay using p-nitrophenyl phosphate as a phosphatase substrate. In order to normalize ALP production in wells with varying cell quantity, ALP quantities measured in each well were divided by the DNA quantity from corresponding wells. Separate plates were fixed with 10% formalin and stained with alizarin red-S (MP Biomedicals) to visualize mineralization microscopically.
Concentrations of each biofilm inhibitor evaluated for osteoblast biocompatibility.
| Biofilm Inhibitor | Concentrations Tested (μM) |
|---|---|
| D-Amino Acid Mixture | 500, 250, 125, 62.5, 31.25 |
| Lysostaphin | 1.5, 0.75, 0.19, 0.094, 0.047 |
| Farnesol | 30,000, 6,000, 1,200, 240, 48 |
| Cis-2-Decenoic Acid | 500, 250, 125, 62.5, 31.25, 15.6 |
| Desformylflustrabromine | 70, 35, 17.5, 8.75 |
Statistical analysis on DNA and ALP content was performed using ANOVA with Tukey post-hoc tests with n = 4 and significance level (α) of 0.05.
RESULTS
Visual Observations of the Chemical Dispersal Test Group Solutions
D-AA mixture formed white flakey precipitates at 500 μM and above, but the lower concentrations were soluble and did not precipitate. The farnesol and C2DA were very hydrophobic and formed micelles, but vigorous shaking created an even distribution of the solute prior to addition to cultures. The remaining agents tested, LS and dFBr, were polar and were therefore soluble and mixed evenly in solution.
Cell Viability
MC3T3 cells in osteogenic media had a positive growth rate until day 3, then cell viability remained steady over the following two weeks as cells became confluent (Fig. 1a).
Osteogenic media containing ethanol had statistically lower viability on day 3 in comparison to the two other control groups, but no significant differences in cell viability between control groups with or without osteogenic additives and ethanol were detected for any other time point. DNA quantity in cells treated with varying concentrations of lysostaphin remained consistent during the study for all concentrations tested (Fig. 1c), suggesting that it is not cytotoxic at concentrations equal to or less than 37.5 µg/mL (1.5 µM). For all evaluated anti-biofilm compounds, except for lysostaphin, there were dose-dependent decreases in viability during the initial 3 days shown in Fig. (1b-f). While cells exposed to C2DA did not exhibit significant decreases in viability by day 1, levels at 62.5 μM and above resulted in slower or inhibited cell growth at day 3 and beyond (Fig. 1e). D-AA initially decreased viability on the first day, but cells recovered and grew, though at decreased rates, at day 3 and beyond up to a concentration of 250 μM (Fig. 1b). Farnesol and dFBr had significant impacts on cell viability at levels over 48 μM (Fig. 1d) and 35 μM respectively (Fig. 1f).
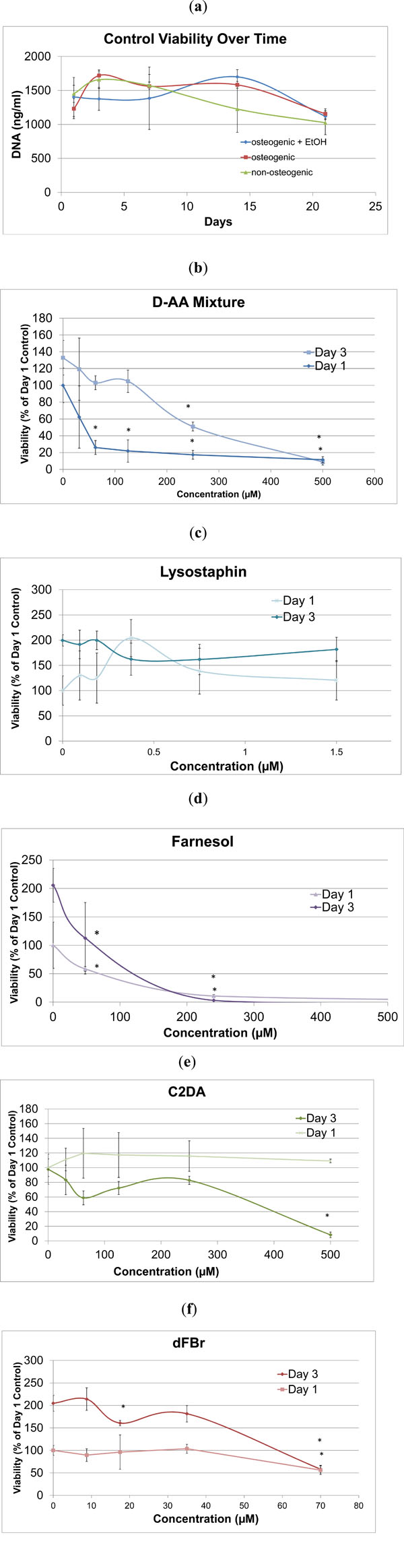
DNA quantity for a) all the control groups and b-f) biofilm inhibitors to osteogenic controls on day 1 and 3 at varying concentrations. Data are represented as mean ± standard deviation; *indicates significant difference compared to osteogenic control.
Visual Observations of Cell Cultures
For focused analysis of mineralization response, the highest concentrations that supported viability were selected for reporting differentiation and mineralization markers (Table 2). In general, biocompatible concentrations of farnesol, D-AA, LS, and C2DA remained confluent and elongated similar to control groups, but dFBr was an exception. dFBr test samples changed morphology from the elongated fibroblast-like spindle shape form to a cuboidal form, similar to that of epithelial cells instead of osteoblast-like cells (Fig. 2h, i) [15]. Significant staining for mineral deposits in osteogenic control groups became apparent by day 14 (Fig. 2b, c).
Comparison of concentration evaluated, reported effective ranges and highest identified sub-toxic concentration.
| Highest Conc. Evaluated (μM) |
Reported Biofilm-Inhibitory Concentration Ranges (μM) |
Highest Sub-Toxic Conc. Evaluated (Shown in Figures) (μM) |
|
|---|---|---|---|
| D-AA | 500 | 10-500 [10, 33] | 250 |
| LS | 1.5 | 0.32-8 [11] | 1.5 (toxic conc. not determined) |
| Farnesol | 240 | 300-3,000 [17, 34] | 48 |
| C2DA | 500 | 0.0025-250 [9, 13] | 62.5 |
| dFBr | 70 | 70-200 [12] | 35 |
The abbreviations stand for the following: N-O (non-osteogenic), O (osteogenic), O+EtOH (osteogenic with 1.25% ethanol), D-AA (D-amino acid), dFBr (desformylflustrabromine), C2DA (cis-2-decenoic acid), and LS (lysostaphin).
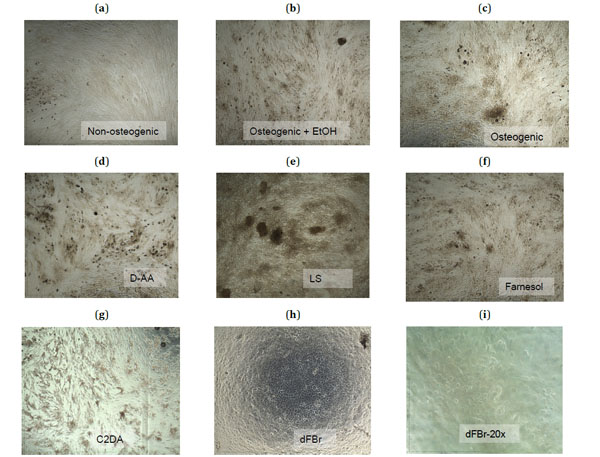
Microscopic images (4X magnification) taken at day 21 of the control groups (a-c) and the highest sub-toxic concentrations of each test group (d-h) stained with Alizarin Red-S to show calcium deposits in dark red-brown. A 20X magnification of cells exposed to dFBr (i) shows the rounded morphology of these cells.
Cell Differentiation
ALP, an early indicator of osteogenic differentiation, peaked around day 3 for the osteogenic and non-osteogenic control groups (Fig. 3a). The osteogenic control group with ethanol had a decreased day 3 ALP production, but had higher ALP/DNA ratios than the other groups after day 3 and continued to positively increase throughout the 21day study. The D-AA test samples decreased in ALP/DNA production over time in comparison to the osteogenic control groups with ethanol (Fig. 3b). No significant differences from the osteogenic control group were detected in the evaluated concentrations of lysostaphin (Fig. 3c). Farnesol stimulated early significant increases in ALP/DNA on day 3 compared to both osteogenic control groups with and without ethanol, then continued to decrease in ALP production throughout the remainder of the 21 day study (Fig. 3d). In a similar fashion to farnesol, the C2DA test group ALP/DNA ratio peaked at day 3 at a significantly higher value than the osteogenic control groups, but steadily declined over the remainder of the 21 day time period (Fig. 3e). The farnesol and C2DA test groups displayed high variability, which may correspond to higher variability in the DNA levels. dFBr significantly lowered ALP production in the early stages of exposure to the cells, but peaked at levels similar to the osteogenic control by day 14 (Fig. 3f). With day 3 representing the peak in control ALP production, only farnesol, C2DA, non-osteogenic media, and osteogenic media containing ethanol sig-nificantly differed from the osteogenic positive control (Fig. 4).
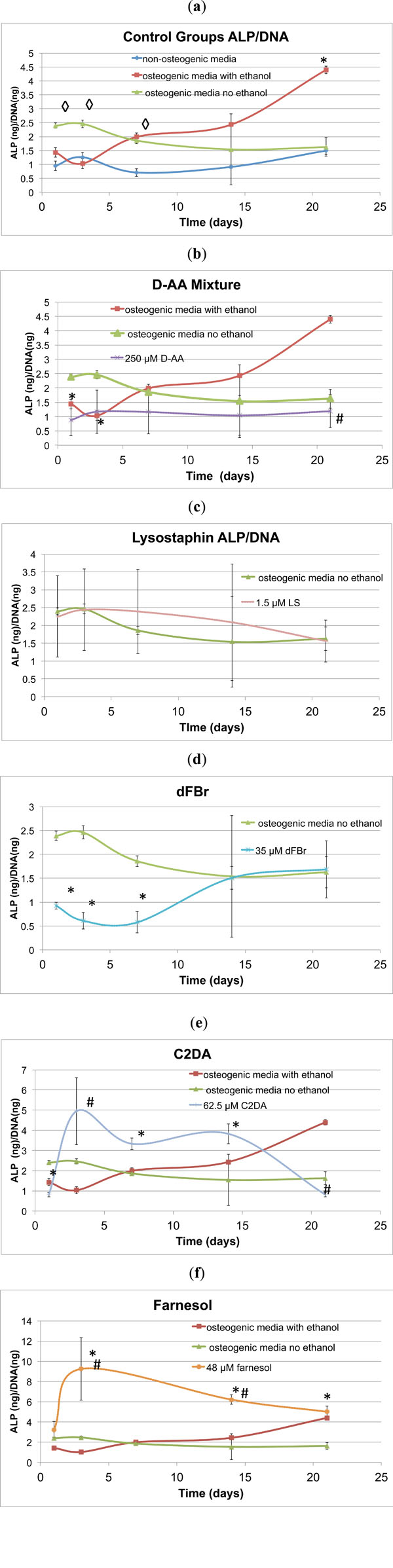
Graphical representations ofALP/DNA ratio over time (mean ± standard deviation) for a) control and b-f) test groups. ◊indicates significant difference compared to non-osteogenic control, * indicates significant difference compared to osteogenic control, # indicates significant difference compared to osteogenic ethanol control.

Graphical representation comparing day 3 ALP/DNA production for control groups and sub-toxic concentrations of test groups. Data are represented as mean ± standard deviation; * indicates significant difference compared to osteogenic control, # indicates significant difference compared to osteogenic ethanol control.
DISCUSSION
The purpose of this study was to identify the biocompatible concentration range of five different biofilm-inhibitory molecules and to identify potential effects on osteoblast function prior to development of clinical applications for local delivery to inhibit orthopaedic infection. All the tested biocompatible concentration ranges were within reported biofilm inhibitory ranges, except farnesol and dFBr. While these anti-biofilm molecules have sub-toxic levels, some may influence osteogenic differentiation by altering ALP production.
The results of this preliminary in vitro study may be interpreted to predict possible in vivo outcomes of these biofilm-inhibitory agents delivered at locally active concentrations. One limitation of this study was the test sample size, n=4, with gapped time points. Samples were taken at time points to reflect early and later stages of differentiation, days 1, 3, 7, 14, and 21 [16]. Increasing the frequency of selected time points may provide more definitive characterization of cell growth and differentiation patterns after exposure to these anti-biofilm agents. The strategy used in this study was repetitive dosing, which applies more stress on the cell samples, as opposed to an approach that involves one initial dose at various concentrations. This strategy was chosen based on an ideal local delivery system, releasing continuous levels over a clinically-relevant time frame. Many local delivery systems display burst response followed by minimal release, which may result in different levels of recovery from the initial higher dose of biofilm-inhibitory chemicals [17]. Other forms of quantitatively measuring differentiation can be used in future studies to further characterize differentiation state by measuring gene expression for osteoblast-specific proteins such as osteocalcin, matrix gla protein, osteopontin, collagen, and bone sialoprotein [18, 19].
The D-AA test group was toxic at high concentrations, but supported cells at intermediate ranges over the course of 21 days with repetitive dosing. This toxic concentration level is much lower than previously reported toxic oral doses of D-AA [20]. D-Proline ingested orally has been shown to increase gastric inflammation, peptic ulcer formation, and kidney fibrosis and necrosis [20]. An equimolar mixture of D-AA was chosen for evaluation in this study based on results by Hochbaum et al. and Sanchez et al. showing optimal efficacy with a combination of various D-AA [10, 21]. Sanchez et al.’s study also evaluated human dermal fibroblasts and osteoblasts exposed to D-AA for 24 hours, showing that cells exposed to 50mM (approximately 100 times the highest concentration evaluated in this study) of D-Methionine, D-Proline, and D-Tryptophan were more than 70% viable. It is important to note for comparison purposes that Sanchez et al.’s study was short term (24 hours), whereas this study was much longer (21 days). Time course, different cell lines, different D-AAs, and solubility enhancement of D-AA in media could contribute to the variation in viability results. The study by Sanchez et al. evaluated anti-biofilm efficacy of released D-AAs in vivo over 14 days, but did not report the quality of bone healing in the segmental defect. The interaction between D-AA and the host is a vital part of the healing process and should be evaluated further in vivo. Finally, the cytotoxicity of individual D-AA could be tested to determine whether a single component is more toxic than others.
There have been other studies involving fatty acid applications for antimicrobial effects against Gram-negative, Gram-positive, fungi, protozoan, and viruses [9, 13, 22]. Fatty acids have high potential as anti-biofilm therapeutics because many are nonspecific to particular types of microorganisms, although they have limitations of instability and a tendency to bind non-specifically to proteins [22]. Similar to a study by Jennings et al. of C2DA, up to 500 μg/mL (3 mM) supported fibroblast viability, but may have affected growth over the course of 48 hours [13]. The present study resulted in cell death at concentrations of C2DA over 62.5 μM by day 3, which continued through the 21 day study. The difference in viability values may be due to the length of the test, 48 hours versus 21 days, or difference of cell lines, fibroblasts versus osteoblasts [13]. However, the lower biocompatibility level this longer term study identified still has the potential to be effective against biofilm. Davies et al. have shown that bacterial colonies can be very sensitive to C2DA, some species in the nanomolar region [9]. There have been several published studies of different cell reactions to the application of various fatty acids to the cultured cells, skin, and teeth in order to inhibit bacterial growth, as well as oral ingestion of fatty acids to control gut microbiota, showing dose-dependent toxicity of different fatty acids [23, 24]. Cell type as well as fatty acid properties may play a role in viability, apoptosis, and necrosis, and effects of fatty acids on other cell types such as endothelial cells, lymphocytes, macrophages, and neutrophils should be studied further [23].
Comparing to results from Unnanuntana et al., concentrations of farnesol that are compatible with osteoblasts are more than six times lower in this study (48 μM compared to 300 μM) [17]. The discrepancy between toxic concentrations found in this study and the study by Unnanuntana et al. could be related to the dosage strategy and the solubility procedure. The reversibility of farnesol effects was studied by exposing cultures to farnesol for two hours, suspended directly in media without additives to improve solubility, after which the farnesol was removed [17]. Previous studies have indicated that the presence of fatty acids in serum can induce osteoblasts to undergo adipogenesis [25]. Masi et al. showed that endothelial cells incubated in fatty acids for 24 hours showed increased fatty deposits above 150 μM, over twice the biocompatible concentrations of farnesol or C2DA [23]. The farnesol, C2DA, and control groups maintained elongated spindle morphology (bone-like). In preliminary studies using oil-red O staining, there was no evidence of fat deposits in cells exposed to C2DA or farnesol compared to controls.
While no previous studies of dFBr in osteoblast culture have been published, it has been studied at concentrations much lower than those used in this study for its ability to interact with specific cell receptors as positive allosteric modulators for potential therapeutic application for various neurological and neuropsychiatric disorders [26]. The chemical structure of dFBr can be modified to develop more selective agents for acetylcholine receptors and potentially biofilm inhibitors. Of the evaluated anti-biofilm agents, dFBr had the most significant effect on osteoblast differentiation activity and morphology. The morphologic change, lack of mineralization, and altered differentiation may have been a result of interaction with specific cell receptors, leading cells to differentiate into an undetermined lineage.
In a growth inhibitory context, LS is effective at concentrations as low as 0.004 µg/mL in some bacterial strains and disrupts S. aureus biofilm at concentrations as low as 0.8 µg/mL. Efficacy in eradicating biofilm in other staphylococcal strains, such as S. epidermidis, has been demonstrated at much higher concentrations of 200 µg/mL. This study only evaluated concentrations within the range effective against S. aureus. However, since the highest concentration of LS was not found to be toxic to osteoblasts in this study, the biocompatibility range could be further explored. This biofilm-disrupting agent may not be toxic within the reported anti-biofilm range because it is an endopeptidase protein that disrupts the cell wall of Staphylococcus aureus with specificity toward Staphylococcal microorganisms [11, 27]. Therefore, LS may be non-toxic over a broad concentration range due to the presumably minimal disruption of mammalian cell membranes. While there have been no published studies evaluating LS effects on osteoblasts, there could be issues of immune reactivity when used in vivo in addition to short protein half-life and specificity toward Staphylococcal strains [27]. These limitations could be overcome by conjugation of these protein agents to polyethylene glycol (PEG), although the effect on biofilm inhibition of PEGylated LS is reported to be slightly decreased [27].
The increased production of ALP in control groups containing ethanol compared to osteogenic media without ethanol, in addition to all of the other test groups, was an unanticipated outcome. Models of chronic ethanol exposure in rats have shown inhibited bone development in vivo and increased proliferation and formation of bone nodules in stem cells isolated from ethanol-exposed rats, though no significant differences in ALP production were observed [28]. Yeh et al. also reported that ethanol reduces osteogenesis in stromal cells isolated from humans with ethanol-induced osteonecrosis, showing that ethanol interferes with the signaling pathways that induced osteoblast formation [29]. In the former studies, chronic alcohol consumption was studied and modeled by isolating cells from animals with ethanol exposure in addition to culture conditions with ethanol at a higher percentage than that used in this study for longer periods of time. An in vitro study by Farley et al. suggests that changes in cell function after ethanol exposure is due to ethanol increasing membrane fluidity [30]. Increased membrane permeability due to ethanol at low doses may have allowed for increased uptake of osteogenic factors. The increased ALP production in the C2DA and farnesol groups was also unexpected. Both of these biofilm inhibitors were delivered in ethanol but were much higher in ALP production than ethanol controls at early time points. This finding may have been an artifact of low cell number or may indicate that proliferative mechanisms have been affected, as several studies have demonstrated an inverse relationship between the sequential processes of proliferation and differentiation [31, 32]. As proliferative mechanisms decreased, ALP production may have begun at an earlier time point in these groups. However, the mineralization staining patterns do not suggest that this led to increased deposition of mineral or differentiation into bone.
In conclusion, in vitro studies of potential biofilm inhibiting/dispersing agents are a vital step in evaluating possible solutions to orthopaedic implant related infections. Several of the tested anti-biofilm agents were biocompatible within its reported effective ranges; however, others were proven cytotoxic within the reported anti-biofilm concentrations. dFBr and farnesol were not biocompatible within the effective anti-biofilm ranges and would require more testing before therapeutic use. High concentrations of naturally produced biofilm inhibitors, such as those that may be present after local delivery, may have effects on cellular viability and osteoblast function. In vivo studies based on dose response limits identified in these studies are needed to fully characterize the effects of these biofilm inhibitors prior to clinical use.
CONFLICT OF INTEREST
The authors confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
ACKNOWLEDGEMENTS
Funding for portions of this research was provided by Department of Defense, Peer Reviewed Orthopaedic Research Program Award # OR090687, and departmental funds at the University of Memphis. The authors also wish to acknowledge Harry Courtney, PhD, from University of Tennessee Health Science Center and Mark Smeltzer, PhD, from University Arkansas for Medical Sciences for advice and assistance in experimental research.

